
Current Research
The Eisenberg Lab applies structural, biochemical, and computational methods to understand the molecular basis of neurodegeneration. Particular projects include the development of small-molecule and peptide-based drugs for Parkinson’s and Alzheimer’s diseases, and the involvement of RNAs in dementias.

In 2022, the Lab used cryo-EM to discover a pharmacophore in the Alzheimer’s-disease-associated fibrils of protein tau. In silico screening into this pharmacophore led to the discovery of small molecules that disassemble the neurotoxic, Alzheimer’s associated fibrils of tau [Reference: “Structure-based discovery of small molecules that disaggregate Alzheimer’s disease tissue derived tau fibrils in vitro,” Paul M. Seidler, Kevin A. Murray, David R. Boyer, Peng Ge, Michael R. Sawaya, Carolyn J. Hu, Xinyi Cheng, Romany Abskharon, Hope Pan, Michael A. De Ture, Christopher K. Williams, Dennis W. Dickson, Harry V. Vinters, David S. Eisenberg, Nature Communications, 13(1):5451 (2022). DOI 10.1038/s41467-022-32951-4. PMID: 36114178. PMCID: PMC9481533.]
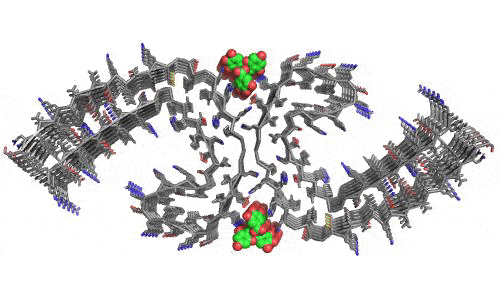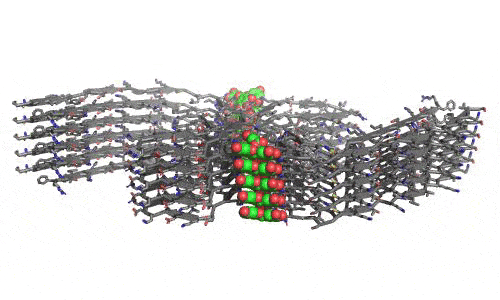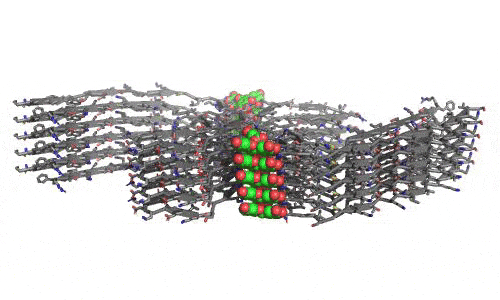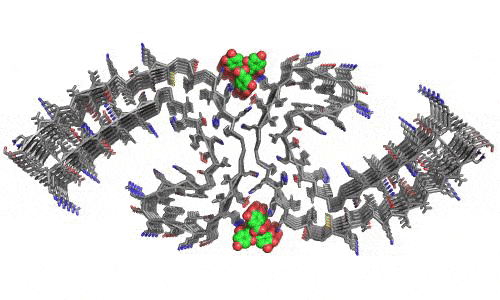
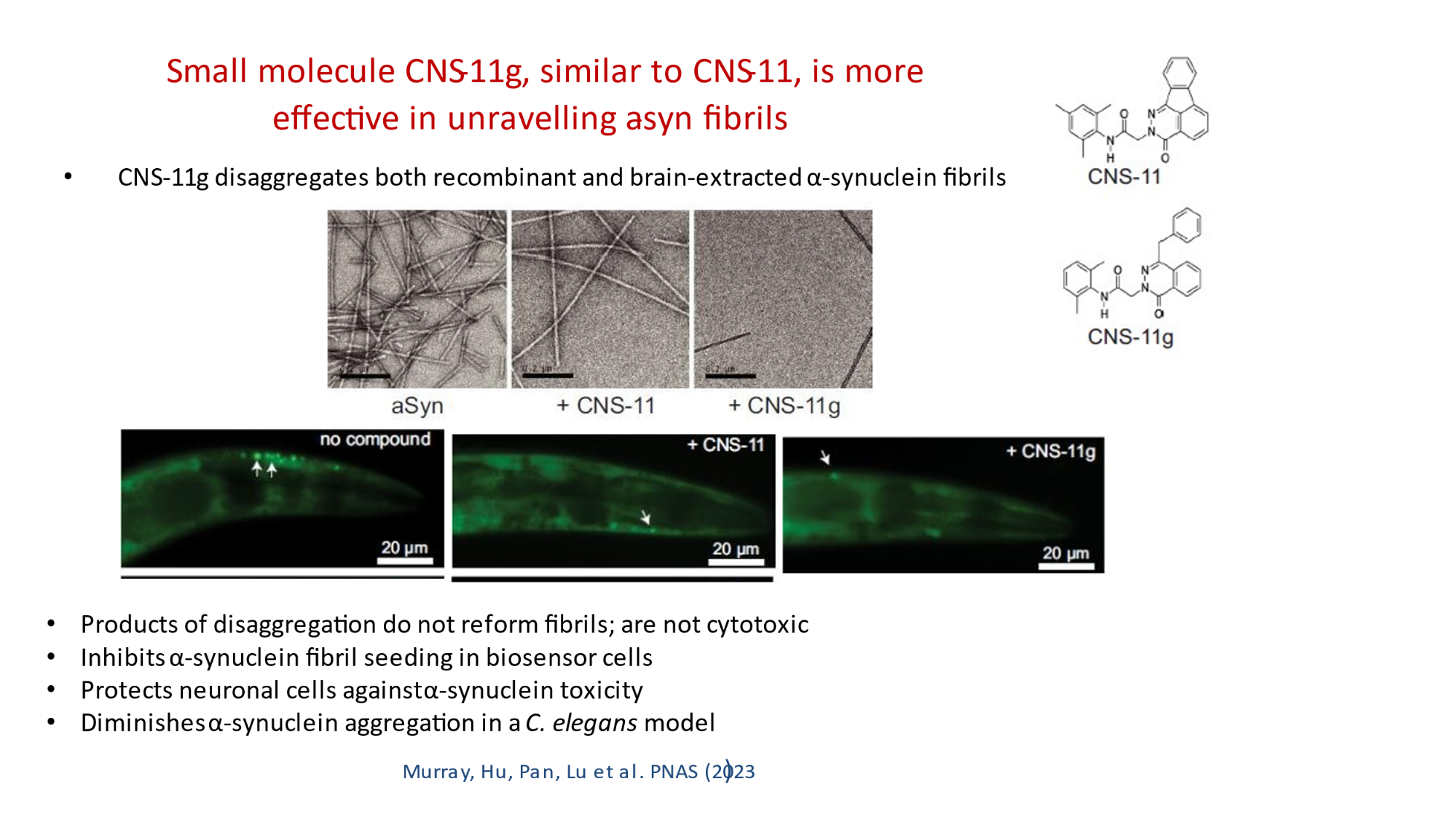
In 2023, the Lab found a related small molecule that disassembled the Parkinson’s-disease-associated fibrils of protein alpha-synuclein, and found it active in C.elegans and mouse models of disease. [Reference: . “Small molecules disaggregate alpha-synuclein and prevent seeding from patient brain-derived fibrils,” Kevin A. Murray, Carolyn J Hu, Hope Pan, Jiahui Lu, Romany Abskharon, Jeannette R. Bowler, Gregory M Rosenberg, Christopher K Williams, Gazmend Elezi, Melinda Balbirnie, Kym F Faull Harry V Vinters, Paul M. Seidler, David S. Eisenberg, Proc Natl Acad Sci USA, Feb (2023), doi: 10.1073/pnas.2217835120, PMCID: PMC9963379.]
In 2024, the Lab reported short peptides that also disassemble Alzheimer’s-disease-associated fibrils of tau. [Reference: D-peptide-magnetic nanoparticles fragment tau fibrils and rescue behavioral deficits in a mouse model of Alzheimer’s disease”, Ke Hou, Hope Pan, Hedieh Shahpasand-Kroner, Carolyn Hu, Romany Abskharon, Paul Seidler, Marisa Mekkittikul, Melinda Balbirnie, Carter Lantz, Michael R. Sawaya, Joshua L. Dolinsky, Mychica Jones, Xiaohong Zuo, Joseph A. Loo, Sally Frautschy, Greg Cole and David S. Eisenberg, Science Advances, 10, 2991 (2024)]
Also in 2024, the Lab presented evidence for the mechanism of fibril disassembly by short peptides. [Reference: “How short peptides can disassemble ultra-stable tau fibrils extracted from Alzheimer’s disease brain by a strain-relief mechanism”, Ke Hou, Peng Ge, Michael R. Sawaya, Joshua L. Dolinsky, Yuan Yang, Yi Xiao Jiang, Liisa Lutter, David R. Boyer, Xinyi Cheng, Justin Pi, Jeffrey Zhang, Jiahui Lu, Shixin Yang, Zhiheng Yu, Juli Feigon, David S. Eisenberg, bioRxiv preprint doi: https://doi.org/10.1101/2024.03.25.586668,*
Biography
David Eisenberg is currently Professor of Chemistry and Biochemistry and Biological Chemistry, as well as HHMI Investigator and Director of the UCLA-DOE Institute for Genomics and Proteomics. Before he came to UCLA, Eisenberg earned an A.B. in Biochemical Sciences from Harvard College and a D.Phil. from Oxford University in Theoretical Chemistry on a Rhodes Scholarship. After postdoctoral study at Princeton University on water and hydrogen bonding and at Caltech on protein crystallography, he joined the faculty at UCLA. Currently he studies protein interactions by X-ray crystallography, bioinformatics, and biochemistry, with an emphasis on amyloid-forming proteins. This recently recognized protein state offers opportunities to understand cells in health and disease, and in synthesizing new materials and in understanding processes as diverse as biofilms and corrosion. Eisenberg has published over 300 papers and reviews, and holds half a dozen patents. His awards include: the UCLA Distinguished Teaching Award, John Simon Guggenheim Fellowship, the UCLA Faculty Research Lectureship, the Stein and Moore Award of the Protein Society, the ACS Faculty Mentoring Award, and membership in the National Academy of Sciences, the American Academy of Arts and Sciences, the American Philosophical Society, and the Institute of Medicine.
Recent Publications
- “Catalytic Synthesis of PEGylated EGCG Conjugates that Disaggregate Alzheimer’s Tau,” Anton El Khoury, Paul M. Seidler, David S. Eisenberg, Patrick G. Harran, Thieme Synthesis 2021; 53(22): 4263-4271 (2021)
- “Prevalence and species distribution of the low-complexity, amyloid-like, reversible, kinked segment (LARKS) structural motif in amyloid-like fibrils,” Hughes MP, Goldschmidt L, Eisenberg DS, J Biol Chem, (2021) PMID: 34537246
- “Cryo-EM structures of hIAPP fibrils seeded by patient-extracted fibrils reveal new polymorphs and conserved fibril cores,” Qin Cao, David R. Boyer, Michael R. Sawaya, Romany Abskharon, Lorena Saelices Binh A. Nguyen, Jiahui Lu, Fouad Kandeel, David S. Eisenberg, Nature Structure and Molecular Biology, September 2021. PMID: 34518699
- “The SARS-CoV-2 nucleocapsid protein preferentially binds long and structured RNAs,” Christen E. Tai, Einav Tayeb-Fligelman, Sarah Griner, Lukasz Salwinski, Jeannette T. Bowler, Romany ABskaron, Xinyi Cheng, Paul M. Seidler, Yi Xiao Jiang, David S. Eisenberg, Feng Guo, bioRxiv, December 2021. https://doi.org/10.1101/2021.12.25.474155
- “Atomic view of an amyloid dodecamer exhibiting selective cellular toxic vulnerability in acute brain slices,” Amber L. H. Gray, Michael R. Sawaya, Debalina Acharyya, Jinchao Lou, Emery M. Edington, Michael D. Best, Rebecca A. Prosser, David S. Eisenberg, Thanh D. Do, Protein Science, 31:716-727. Dec 26 2021 ISSN: 0961-8368 , 1469-896X; DOI: 10.1002/pro.4268. PMID: 3495485. PMCID: PMC8862425.
- “Bioinformatic identification of previously unrecognized amyloidogenic proteins,” Gregory M. Rosenberg, Kevin A. Murray, Lukasz Salwinski, Michael P. Hughes, Romany Abskharon, David S. Eisenberg, JBC, April 9, 2022, https://doi.org/10.1016/j.jbc.2022.101920
- “Cryo-EM structure of RNA-induced tau fibrils reveals a small C-terminal core that may nucleate fibril formation,” Romany Abskharon, Michael R Sawaya, David R Boyer, Qin Cao, Binh A Nguyen, Duilio Cascio, David S Eisenberg, PNAS USA, Apr 12 2022; 119 (15):e2119952119. doi: 10.1073/pnas.2119952119. PMID: 35377792
- “Amyloid fibrils in disease FTLD-TDP are composed of TMEM106B not TDP-43,” Yi Xiao Jiang, Zin Cao, Michael R. Sawaya, Romany Abskharon, Peng Ge, Michael DeTure, Dennis W. Dickson, Janine Y. Fu, Rachel R Ogorzalek Loo, Joseph A Loo, David S Eisenberg, Nature, March 28 2022, 605:304-309. PMID: 35344984
- “Identifying amyloid-related diseases by mapping mutations in low-complexity protein domains to pathologies,” David S. Eisenberg, Kevin A. Murray, Michael P. Hughes, Carolyn J. Hu, Michael R. Sawaya, Lukasz Salwinski, Hope Pan, Samuel W. French, Paul M. Seidler, NSMB, 29(6):529-536. Jun 2022 May 30. doi:10.1038/s41594-022-00774-y. PMID: 35637421. PMCID: PMC9205782A381.
- “The rippled Beta-sheet layer configuration- a novel supramolecular architecture based on predictions by Pauling and Corey,” Amaruka Hazari, Michael R. Sawaya, Niko Vlahakis, Timothy C. Johnstone, David Boyer, Jose Rodriguez, David Eisenberg, and Jevgenji A. Raskatov, Chem Sci, July 2022, 13:8947-8952, https://doi.org/10.1039/D2SC02531K
- “De novo designed protein inhibitors of amyloid aggregation and seeding,” Kevin A. Murray, Carolyn J. Hu, Sarah L. Griner, David S. Eisenberg, Biophysics and Computational Biology, August 15, 2022, https://doi.org/10.1073/pnas.2206240119
- “Structure-based discovery of small molecules that disaggregate Alzheimer’s disease tissue derived tau fibrils in vitro,” Paul M. Seidler, Kevin A. Murray, David R. Boyer, Peng Ge, Michael R. Sawaya, Carolyn J. Hu, Xinyi Cheng, Romany Abskharon, Hope Pan, Michael A. De Ture, Christopher K. Williams, Dennis W. Dickson, Harry V. Vinters, David S. Eisenberg, Nature Communications, 13(1):5451 (2022). DOI 10.1038/s41467-022-32951-4. PMID: 36114178. PMCID: PMC9481533.
- “Micro-electron diffraction structure of the aggregation-driving N-terminus of Drosophila neuronal protein Orb2A reveals amyloid-like b-sheets,” Jeanette Bowler, Michael R. Sawaya, David R. Boyer, Duilio Cascio, Manya Bali, David S. Eisenberg, Journal of Biological Chemistry, ms. #JBC-D-22-00423R2 . J Biol Chem. 2022 Oct;298(10):102396. doi: 10.1016/j.jbc.2022.102396. Epub 2022 Aug 18. PMID: 35988647. PMCID: PMC9556795
- “Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee,” Joel N. Buxbaum, Angela Dispenzieri, David S. Eisenberg, Marcu Fanrich, Giampaolo Merlini, Maria J.M. Saraiva, Yoshiki Sekijima and Per Westermark, Amyloid, Dec 2022; 29(4): 213-219, doi: 10.1080/13506129.2022.2147636. Epub 2022 Nov 24.
- “Small molecules disaggregate alpha-synuclein and prevent seeding from patient brain-derived fibrils,” Kevin A. Murray, Carolyn J Hu, Hope Pan, Jiahui Lu, Romany Abskharon, Jeannette R. Bowler, Gregory M Rosenberg, Christopher K Williams, Gazmend Elezi, Melinda Balbirnie, Kym F Faull Harry V Vinters, Paul M. Seidler, David S. Eisenberg, Proc Natl Acad Sci USA, Feb (2023), doi: 10.1073/pnas.2217835120, PMCID: PMC9963379.
- “DNAJB8 oligomerization is mediated by an aromatic-rich motif that is dispensable for substrate activity,” Bryan D Ryder, David R Boyer, Elizaveta Ustyantseva, Ayde Mendoza-Oliva, Mikolaj I Kuska, Pawel M Wydorski, Michael Sawaya, Marc Diamond, David S Eisenberg, Harm H Kampinga, Lukasz A Joachimiak, bioRxiv, March 2023, doi: 10.1101/2023.03.05.531355. Preprint. PMCID: PMC10028812
- “Low complexity domains of the nucleocapsid protein of SARS-CoV-2 form amyloid fibrils,” Einav Tayeb-Fligelman, Jeannette T Bowler, Christen E Tai, Michael R Sawaya, Yi Xiao Jiang, Gustavo Garcia Jr, Sarah L Griner, Xinyi Cheng, Lukasz Salwinski, Liisa Lutter, Paul M Seidler, Jiahui Lu, Gregory M Rosenberg, Ke Hou, Romany Abskaron, Hope Pan, Chih-Te Zee, David R Boyer, Yan Li, Danel H Anderson, Kevin A Murray, Genesis Falcon, Duilio Cascio, Lorena Saelices, Robert Damoiseaux, Vaithilingaraja Arumugaswami, Feng Guo, David S Eisenberg, Nature Communications, April 2023, doi: 10.1038/s41467-023-37865-3. PMCID: PMC10127185
- “Structure-based design of nanobodies that inhibit seeding of Alzheimer’s patient-extracted tau fibrils.” Abskharon, R, Pan H, Sawaya MR, Seidler PM, Olivares, EJ, Chen Y, Murray KA, Zhang J, Lantz C, Bentzel M, Boyer DR, Cascio D, Nguyen BA, Hou K, Cheng X, Parton E, Williams CK, Nana AL, Vinters HV, Spina S, Grinberg LT, Seeley WW, Steyaert J, Glabe CG, Loo RO, Loo JA, Eisenberg D. PNAS. October 2023. 120:41, e2300258120; doi: 10.1073/pnas.2300258120. PMCID: PMC10576031
- “Fibril structures of TFG protein mutants validate identification of TFG as a disease-related amyloid protein by the IMPAcT method,” Gregory M. Rosenberg, Romany Abskharon, David R. Boyer, Peng Ge, Michael R. Sawaya, David S. Eisenberg, PNAS Nexus, 2, 1-14 (2023) PMID: 38077690,.
- “Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy”, Nguyen BA, Singh V, Afrin S, Yakubovska A, Wang L, Ahmed Y, Pedretti R, Fernandez-Ramirez MDC, Singh P, Pekala M, Cabrera Hernandez LO, Kumar S, Lemoff A, Gonzalez-Prieto R, Sawaya MR, Eisenberg DS, Benson MD, Saelices L. Nat Commun. 2024 Jan 1; 15(1):581 2024 PMID: 38233397
- “Cryo-EM structures of the D290V mutant of the hnRNPA2 low-complexity domain suggests how D290V affects phase separation and aggregation“, Lu J, Ge P, Sawaya MR, Hughes MP, Boyer DR, Cao Q, Abskharon R, Cascio D, Tayeb-Fligelman E., Eisenberg DE. J Biol Chem. (2024) Feb; 300(2):105531. Epub 2023 Dec 9. 2024 PMID: 38072051
- “D-peptide-magnetic nanoparticles fragment tau fibrils and rescue behavioral deficits in a mouse model of Alzheimer’s disease”, Ke Hou, Hope Pan, Hedieh Shahpasand-Kroner, Carolyn Hu, Romany Abskharon, Paul Seidler, Marisa Mekkittikul, Melinda Balbirnie, Carter Lantz, Michael R. Sawaya, Joshua L. Dolinsky, Mychica Jones, Xiaohong Zuo, Joseph A. Loo, Sally Frautschy, Greg Cole and David S. Eisenberg, Science Advances, 10, 2991 (2024) PMID: 38691615
- “How short peptides can disassemble ultra-stable tau fibrils extracted from Alzheimer’s disease brain by a strain-relief mechanism”, Ke Hou, Peng Ge, Michael R. Sawaya, Joshua L. Dolinsky, Yuan Yang, Yi Xiao Jiang, Liisa Lutter, David R. Boyer, Xinyi Cheng, Justin Pi, Jeffrey Zhang, Jiahui Lu, Shixin Yang, Zhiheng Yu, Juli Feigon, David S. Eisenberg, bioRxiv doi: 10.1101/2024.03.25.586668
- “Liganded magnetic nanoparticles for magnetic resonance imaging of α-synuclein”, Hope Pan, Melinda Balbirnie, Ke Hou, Naomi S. Sta Maria, Shruti Sahay, Paul Denver, Stefano Lepore, Mychica Jones, Xiaohong Zuo, Chunni Zhu, Hilda Mirbaha, Hedieh Shahpasand-Kroner, Marisa Mekkittikul, Jiahui Lu, Carolyn Hu, Xinyi Cheng, Romany Abskharon, Michael R. Sawaya, Christopher K. Williams, Harry V. Vinters, Russell E. Jacobs, Neil G. Harris, Gregory M. Cole, Sally A. Frautschy, David S. Eisenberg, Submitted, Parkinson’s Disease, 2024
- “Structure-based discovery of small molecules that disaggregate tau fibrils in a mouse model of tauopathy”, Hope Pan, Xinyi Cheng, Jeffrey Zhang, Ke Hou, Kapil Manglani, Cansheng Zhu, Marisa Mekkittikul, Tyler Halladay, Hilda Mirbaha, Gazmend Elezi, Romany Abskharon, Michael R. Sawaya, Alexander Bombino, Christopher K. Williams, Harry V. Vinters, Julian P. Whitelegge, Gregory M. Cole, Sally A. Frautschy, David S. Eisenberg, in preparation.
2020-2021 Lab Picture

2015 Lab Picture
