Structural Effects of Amyloidogenic Mutations
For a PDF version of this document: Download this file.
Please see our accompanying review Genetic and structural aspects of amyloid diseases, Sci. Transl. Med., Vol 17, Issue 818, Oct. 1 2025
When atomic structures are known for both the amyloid fibrils formed by the wild-type protein and for a mutational variant of it, comparison of the two structures can reveal how the structural changes contribute to the conversion to the amyloid state. Of the proteins which are known to form amyloid in both wild-type and mutant forms, there are known structures for both the wild-type fibril and the mutant fibril for 10 proteins. In the associated review article, we describe the structural basis for mutation-driven conversion to the amyloid state of amyloid-β, heterogeneous nuclear ribonucleoprotein A2, and transthyretin. Here we describe the seven other cases: Cytotoxic granule associate RNA binding protein TIA1, islet amyloid polypeptide, major prion protein, microtubule-associated protein tau, TAR DNA-binding protein 43, α-synuclein, and β2-microglobulin.
Note, there are many existing fibril structures which are not reviewed here. The structures highlighted here are chosen for the sake of informative comparison between mutant and wild-type structures as well as biological relevance. For this purpose, we aim to compare wild-type and mutant structures which are most similar to one another so that we can elucidate the minimal structural changes which the mutation is sufficient to induce. In addition, where available, patient-derived structures are prioritized for comparison.
Cytotoxic granule associated RNA binding protein TIA1 ↓
Cytotoxic granule associated RNA binding protein TIA1, or just RNA-binding protein TIA1, is a protein which forms functional amyloid fibrils which play a role in the formation and function of stress granules(Rayman and Kandel 2017). This protein engages in reversible aggregation via its prion-like domain (PLD) which is the site of many of its pathogenic mutations linked to ALS and Welander distal myopathy (WDM). The mutation for which we have structural information is G355R. Unlike other mutations in this protein, the G355R mutation reduces fibril stability and delays amyloid aggregation(Inaoka et al. 2023). Since RNA-binding protein TIA1 is a functional amyloid, this reduction in amyloidogenic behavior constitutes a kind of loss-of-function mechanism. Even so, the protein is still able to form amyloid fibrils while harboring the mutation, so these nonfunctional fibrils are ostensibly pathological amyloid fibrils. The molecular structures of the wild-type (pdb ID: 8j3k) and mutant (pdb ID: 8j6c) fibrils, both determined in the same publication(Inaoka et al. 2023) from recombinant constructs of part of the PLD (residues 320-386), reveal the changes which cause the mutant fibrils’ dysfunction.
The wild-type fibril core has two identical protofilaments which each contain a kinked segment similar to low-complexity, aromatic-rich, kinked segments (LARKS)(Hughes et al. 2018), a segment of polar residues participating in a polar zipper, and a segment with several proline residues. These features may contribute to the reversibility of the fibril. The G355R mutation, which would be situated in the proline-rich segment, leads to the formation of a fibril core which completely lacks that proline-rich segment, so the mutant residue is not resolved in the fibril structure. This may be due to the large arginine side chain disrupting the tight conformation of the wild-type glycine observed in the wild-type structure. While the wild-type fibril core has a polar zipper at the center, the mutant fibril core has a kinked segment (LARKS), although with a different conformation than the wild-type structure, and also has a residue in the conformation of an extended amyloid-like low-complexity segment (EAGLS)(Murray et al. 2022). These features of the mutant fibril make it much less stable than the already weakly stable wild-type fibril. This disparity is further evidenced by MD/3D-RISM calculations of stabilization energy of the fibrils which showed that the G355R mutant fibril had less favorable stabilization energy than the wild-type(Inaoka et al. 2023).
Hughes, Michael P., Michael R. Sawaya, David R. Boyer, Lukasz Goldschmidt, Jose A. Rodriguez, Duilio Cascio, Lisa Chong, Tamir Gonen, and David S. Eisenberg. 2018. “Atomic Structures of Low-Complexity Protein Segments Reveal Kinked β Sheets That Assemble Networks.” Science (New York, N.Y.) 359 (6376): 698–701. https://doi.org/10.1126/science.aan6398.
Inaoka, Daigo, Tomoko Miyata, Fumiaki Makino, Yasuko Ohtani, Miu Ekari, Ryoga Kobayashi, Kayo Imamura, et al. 2023. “ALS-Associated Mutation Disturbs Amyloid Fibril Formation of TIA-1 Prion-like Domain.” Preprint. In Review. https://doi.org/10.21203/rs.3.rs-2950744/v1.
Murray, Kevin A., Declan Evans, Michael P. Hughes, Michael R. Sawaya, Carolyn J. Hu, Kendall N. Houk, and David Eisenberg. 2022. “Extended β-Strands Contribute to Reversible Amyloid Formation.” ACS Nano 16 (2): 2154–63. https://doi.org/10.1021/acsnano.1c08043.
Rayman, Joseph B., and Eric R. Kandel. 2017. “TIA-1 Is a Functional Prion-Like Protein.” Cold Spring Harbor Perspectives in Biology 9 (5): a030718. https://doi.org/10.1101/cshperspect.a030718.
Islet amyloid polypeptide ↓
Islet amyloid polypeptide, also called amylin, is a peptide hormone secreted by β-cells of the pancreatic islets of Langerhans which, like other peptide hormones, is resultant from the processing of a larger prohormone. Its physiological function is not entirely clear, but its main function seems to be related to regulation of glucose metabolism by modifying the activity of insulin(Per Westermark, Andersson, and Westermark 2011). Aggregation of this protein has been linked to type II diabetes and it is the main component of islet amyloid deposits in type II diabetes patients(Per Westermark, Andersson, and Westermark 2011; P. Westermark et al. 1986; 1987). A mutation in the IAPP gene, S20G (the numbering for the unprocessed prohormone is S53G), is associated with early onset type 2 non-insulin dependent diabetes(Sakagashira et al. 1996). Biochemical assays on this mutation show that the mutant protein is more amyloidogenic than the wild-type(Ma et al. 2001; Sakagashira et al. 2000; Meier et al. 2016) and it has been demonstrated that a possible mechanism is a decrease in the entropy cost of fibril formation by the mutation increasing local flexibility to encourage certain long-range interactions within the peptide that “preorganize” it toward fibril formation(Xu, Jiang, and Mu 2009).
All extant structures of islet amyloid polypeptide fibrils are from constructs which are synthetic, recombinant, or synthetic but seeded by patient-extracted fibrils. For the purposes of this review, we will focus on a comparison of a wild-type (pdb ID: 6zrf) and two S20G structures (pdb IDs: 6zrq,6zrr) from synthetic peptides reported together in the one publication(Gallardo et al. 2020) (Supp. Fig. 1). The protofilament common to both mutant structures has a bend at position 20 which turns the backbone in the opposite direction of the backbone of the wild-type protofilament, causing some of the side chains which are buried in the wild-type structure to become solvent-facing and vice versa (Supp. Fig. 1a,c). This structural change also alters which residues participate in the protofilament interface.
The consequences of these structural changes are not entirely clear, but the authors report that the wild-type fibril structure is the most stable out of their three structures, even compared to the mutant structure with 3 protofilaments. The observation that the mutant protein aggregates more rapidly, but forms a less stable fibril is consistent with the idea that the mutation drops the entropic cost of the monomer to transition to a fibril by altering the preferred fold of the monomer rather than driving the protein toward a more stable fibril structure. Therefore the mechanism by which the mutation encourages amyloidogenicity seems to be native structure destabilization.
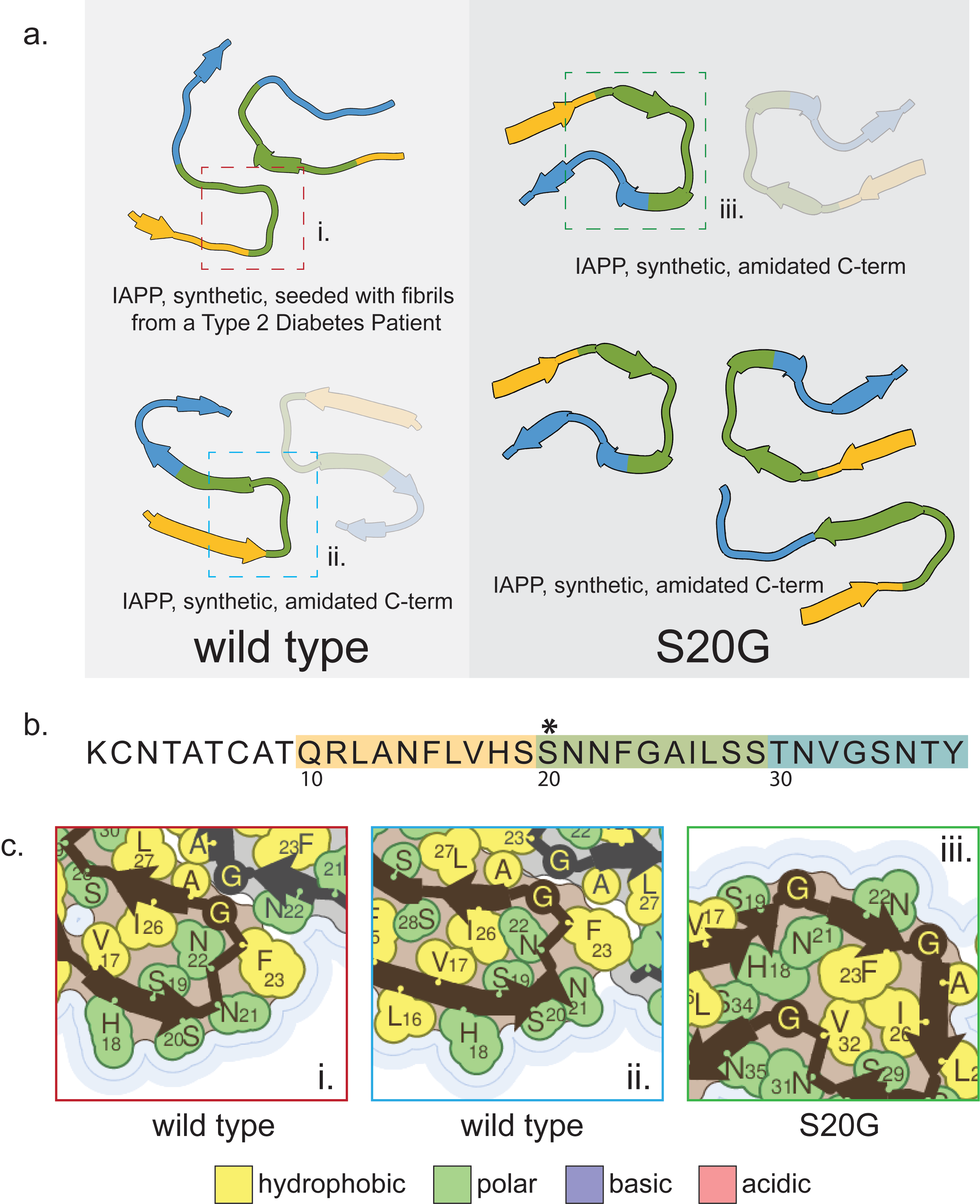
Supplemental Figure 1: Fibril structures of islet amyloid polypeptide. a) Structures of amyloid fibril cores of wild-type (top: pdb id 7m61; bottom: pdb id 6zrf) and S20G (top: pdb id 6zrq; bottom: pdb id 6zrr) islet amyloid polypeptide. b) Amino acid sequence of islet amyloid polypeptide represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). Mutated residues are highlighted by a red outline.
Gallardo, Rodrigo, Matthew G. Iadanza, Yong Xu, George R. Heath, Richard Foster, Sheena E. Radford, and Neil A. Ranson. 2020. “Fibril Structures of Diabetes-Related Amylin Variants Reveal a Basis for Surface-Templated Assembly.” Nature Structural & Molecular Biology 27 (11): 1048–56. https://doi.org/10.1038/s41594-020-0496-3.
Ma, Z., G. T. Westermark, S. Sakagashira, T. Sanke, A. Gustavsson, H. Sakamoto, U. Engström, K. Nanjo, and P. Westermark. 2001. “Enhanced in Vitro Production of Amyloid-like Fibrils from Mutant (S20G) Islet Amyloid Polypeptide.” Amyloid: The International Journal of Experimental and Clinical Investigation: The Official Journal of the International Society of Amyloidosis 8 (4): 242–49. https://doi.org/10.3109/13506120108993820.
Meier, Daniel T., Leon Entrup, Andrew T. Templin, Meghan F. Hogan, Mahnaz Mellati, Sakeneh Zraika, Rebecca L. Hull, and Steven E. Kahn. 2016. “The S20G Substitution in hIAPP Is More Amyloidogenic and Cytotoxic than Wild-Type hIAPP in Mouse Islets.” Diabetologia 59 (10): 2166–71. https://doi.org/10.1007/s00125-016-4045-x.
Sakagashira, S., H. J. Hiddinga, K. Tateishi, T. Sanke, T. Hanabusa, K. Nanjo, and N. L. Eberhardt. 2000. “S20G Mutant Amylin Exhibits Increased in Vitro Amyloidogenicity and Increased Intracellular Cytotoxicity Compared to Wild-Type Amylin.” The American Journal of Pathology 157 (6): 2101–9. https://doi.org/10.1016/S0002-9440(10)64848-1.
Sakagashira, S., T. Sanke, T. Hanabusa, H. Shimomura, S. Ohagi, K. Y. Kumagaye, K. Nakajima, and K. Nanjo. 1996. “Missense Mutation of Amylin Gene (S20G) in Japanese NIDDM Patients.” Diabetes 45 (9): 1279–81. https://doi.org/10.2337/diab.45.9.1279.
Westermark, P., C. Wernstedt, E. Wilander, D. W. Hayden, T. D. O’Brien, and K. H. Johnson. 1987. “Amyloid Fibrils in Human Insulinoma and Islets of Langerhans of the Diabetic Cat Are Derived from a Neuropeptide-like Protein Also Present in Normal Islet Cells.” Proceedings of the National Academy of Sciences of the United States of America 84 (11): 3881–85. https://doi.org/10.1073/pnas.84.11.3881.
Westermark, P., C. Wernstedt, E. Wilander, and K. Sletten. 1986. “A Novel Peptide in the Calcitonin Gene Related Peptide Family as an Amyloid Fibril Protein in the Endocrine Pancreas.” Biochemical and Biophysical Research Communications 140 (3): 827–31. https://doi.org/10.1016/0006-291x(86)90708-4.
Westermark, Per, Arne Andersson, and Gunilla T. Westermark. 2011. “Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus.” Physiological Reviews 91 (3): 795–826. https://doi.org/10.1152/physrev.00042.2009.
Xu, Weixin, Ping Jiang, and Yuguang Mu. 2009. “Conformation Preorganization: Effects of S20G Mutation on the Structure of Human Islet Amyloid Polypeptide Segment.” The Journal of Physical Chemistry. B 113 (20): 7308–14. https://doi.org/10.1021/jp8106827.
Major Prion Protein ↓
Major prion protein is the infectious agent of human prion diseases as well as the subject of mutations which cause inherited human prion diseases. Part of the protein experiences a transition from α-helical structure to β-sheet(Pan et al. 1993), caused by templating on an already misfolded copy of the protein (infection) or destabilization induced by mutations. The mechanism of sporadic cases of prion disease (sporadic Creutzfeldt-Jakob disease, in particular) are not clear, but may be due to somatic mutations or spontaneous conversion of the protein into the amyloidogenic form(Prusiner 1998), but there are risk factors such as homozygosity at codon 129 (rs1799990)(Mead et al. 2012; Palmer et al. 1991).
There are over 60 known mutations in the major prion protein gene (PRNP), but ~85% of hereditary prion diseases are caused by five of them: E200K, V210I, V180I, D178N, and P102L(Minikel et al. 2016). Three mutations have their associated amyloid structures determined, but they are not any of the five listed above. The mutations with solved structures are E196K (pdb ID: 7dwv)(Wang et al. 2021), Y145* (pdb ID: 7rl4)(Li, Jaroniec, and Surewicz 2022), and F198S (pdb IDs: 7umq,7un5)(Hallinan et al. 2022) (Supp. Fig. 2). Only the F198S structure is from patient-derived fibrils. It is difficult to directly compare all these structures to the same wild-type structure since each was formed from a different fragment and they each have different portions of the protein sequence resolved. So, to compare the structures which are the most similar in the resolved region of the fibril core, the F198S structure and Y145X structure will be compared to the wild-type structure published by Glynn et. al. (pdb ID: 6uur)(Glynn et al. 2020) and the E196K structure will be compared to the wild-type structure published by Wang et. al. (pdb ID: 6lni)(Wang et al. 2020) (Supp. Fig. 2). Another difficulty is that only the E196K structure has the actual mutation site resolved in the structure.
Residue 198 is not actually resolved in the F198S structure, so its influence is difficult to discern on the overall structure, but there is another sequence difference between it and the Glynn wild-type structure: the polymorphism at residue 129. Patients with the F198S mutation who are homozygous for valine rather than methionine at position 129 generally have earlier disease onset than those heterozygous at that position, and in the patient from which the amyloid fibrils were extracted F198S was inherited in cis with valine 129(Hallinan et al. 2022). The F198S structure is sterically incompatible with a methionine at position 129 because the valine is packed too tightly (Supp. Fig. 2c). Even with fibrils extracted from M/V heterozygous individuals, the electron density map was more consistent with valine rather than methionine at position 129, indicating that M129 prion protein was less likely to be incorporated into the fibrils. However, the protein is clearly able to form fibrils with a methionine in this position as demonstrated by the wild-type structures from Glynn and Wang as well as the Y145* structure from Li. So, residue 129 is probably exerting influence over which fibril structure forms. But the familial mutation is at position 198, so what effect is that having on the structure? In fact, the proteins making up the aggregates in Gerstmann–Sträussler–Scheinker disease are proteolytically cleaved and do not even retain residue 198(Tagliavini et al. 1991). The group reporting the structure performed mass spectrometry on the brain-extracted protein and found fragments corresponding to the full length protein without N- and C-terminal signal peptides, but the majority of observed peptides span approximately from G80 to V160(Hallinan et al. 2022). The most consistent explanation is that the F198S mutation is affecting the native structure such that it is aberrantly processed and cleaved into amyloidogenic fragments, which could be a result of increased flexibility due to the mutation(Lee et al. 2010). So, the amyloidogenic mechanisms of this mutation are probably native structure destabilization and altered processing. The valine at position 129 acts as a disease modifier by preferring an amyloid fold which is overall more stable than the wild-type M129 fold (based on solvation energy calculations(Sawaya et al. 2021)), but unable to incorporate monomers with a methionine at position 129. Homozygosity at position 129 more consistently results in the production of fibrils with this more stable fold. Interestingly, the F198S structure of major prion protein is sterically incompatible with other known familial mutations for GSS, such as G131V, implying that individuals carrying other mutations in PRNP may harbor distinct fibril structures, yet manifest the same disease.
Unlike the F1298S structure, the Y145* structure has a methionine at position 129, so can be more directly compared to the Glynn wild-type structure (Supp. Fig. 2). However, the mutation is a truncation, and while the wild-type structure was solved from a recombinant protein spanning residues 94-178, the structure only resolves residues 106-145, and Y145 is facing solvent and not necessarily as important for the formation of that particular structure as the electrostatic interactions formed by D144 (Supp. Fig. 2c). In the same vein, the Y145* structure, solved from a recombinant protein spanning residues 23-144, does not resolve the truncated C-terminal, starting at residue N108 and stopping at residue F141. Since the wild-type and mutant structures resolve very similar portions of the protein despite being formed from quite different constructs, it is difficult to explain why the structures are so vastly different. The structural differences may simply be a result of the different methods of generating the fibrils in each case: the mutant fibrils were grown under quiescent conditions at 400μM concentration for a week, sonicated, then used to seed a diluted sample (40μM) under quiescent conditions, and sonicated again; the wild-type fibrils were grown at ~100μM with acoustic resonance mixing for 1-3 days. The buffers in which the fibrils were grown were also quite different from each other with the wild-type fibrils being grown in a slightly denaturing buffer at pH 4, while the mutant fibrils were grown in non-denaturing buffer at pH 6.5. These differing growth conditions may do more to explain the differences in observed structures than the different starting protein segments, given that the portion of the protein which ended up in the rigid fibril core was relatively consistent between the two constructs.
The E196K structure and the wild-type structure spanning residues 170-229 were formed under identical fibrillization conditions to one another and reported by the same group, so their direct comparison has fewer caveats and is more justified (Supp. Fig. 2). The mutant structure also contains the mutant residue, so its effects are more observable. The mutation flips the charge of residue 196 from negative to positive, which disrupts an important interaction between the protofilaments in the wild-type structure: an electrostatic interaction between E196 and K194 (Supp. Fig. 2c). Due to this, an overall rearrangement of the fibril core occurs to form new electrostatic interactions: K196 now interacts with E200 and K194 now interacts with E207 (Supp. Fig. 2c). This change induces other differences including a hydrophilic cavity in the mutant structure instead of a hydrophobic one in the wild-type, additional hydrophobic interactions between the two protofilaments in the mutant structure, and an increase in β-sheet content in the mutant. However, chemical denaturation and thermostability assays indicated that the mutant fibrils were actually less stable than the wild-type fibrils. It has been suggested that the effect of the mutation is destabilization of the monomer through disruption of important salt bridges, which accelerates fibrillization(Hadži et al. 2015), so the amyloidogenic mechanism of this mutation seems to be native structure destabilization. The structural differences of the amyloid fibrils are, therefore, more comparable to strain differences between individuals with prion diseases. Here, the structure is just one piece of the puzzle regarding disease progression variation observed as a function of mutation.
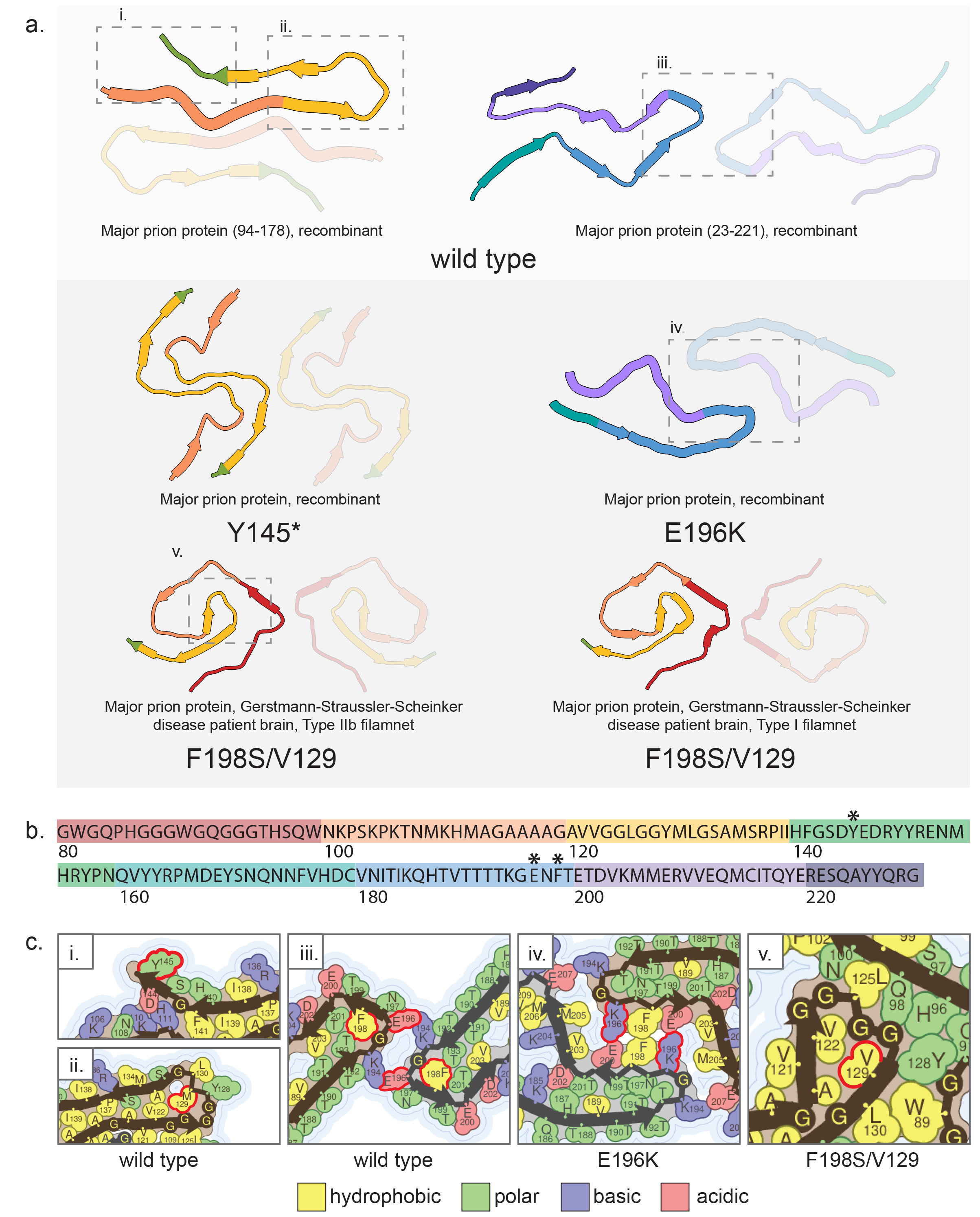
Supplemental Figure 2: Fibril structures of major prion protein. a) Structures of amyloid fibril cores of wild-type (left: pdb id 6uur; right: pdb id 6lni), Y145* (pdb id 7rl4), F198S/V129 (left: pdb id 7un5; right: pdb id 7umq), and E196K (pdb id 7dwv) major prion protein. b) Amino acid sequence of major prion protein represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). Mutated residues are highlighted by a red outline. In this case, for the pdb id 6uur wild-type structure (box ii) and the F198S/V129 mutant structure (box v), residue 129 is highlighted despite being a polymorphism because it is relevant to the structural differences.
Glynn, Calina, Michael R. Sawaya, Peng Ge, Marcus Gallagher-Jones, Connor W. Short, Ronquiajah Bowman, Marcin Apostol, Z. Hong Zhou, David Eisenberg, and Jose A. Rodriguez. 2020. “Cryo-EM Structure of a Human Prion Fibril with a Hydrophobic, Protease-Resistant Core.” Nature Structural & Molecular Biology 27 (5): 417–23. https://doi.org/10.1038/s41594-020-0403-y.
Hadži, San, Andrej Ondračka, Roman Jerala, and Iva Hafner-Bratkovič. 2015. “Pathological Mutations H187R and E196K Facilitate Subdomain Separation and Prion Protein Conversion by Destabilization of the Native Structure.” FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 29 (3): 882–93. https://doi.org/10.1096/fj.14-255646.
Hallinan, Grace I., Kadir A. Ozcan, Md Rejaul Hoq, Laura Cracco, Frank S. Vago, Sakshibeedu R. Bharath, Daoyi Li, et al. 2022. “Cryo-EM Structures of Prion Protein Filaments from Gerstmann-Sträussler-Scheinker Disease.” Acta Neuropathologica 144 (3): 509–20. https://doi.org/10.1007/s00401-022-02461-0.
Lee, Seungjoo, Lizamma Antony, Rune Hartmann, Karen J. Knaus, Krystyna Surewicz, Witold K. Surewicz, and Vivien C. Yee. 2010. “Conformational Diversity in Prion Protein Variants Influences Intermolecular Beta-Sheet Formation.” The EMBO Journal 29 (1): 251–62. https://doi.org/10.1038/emboj.2009.333.
Li, Qiuye, Christopher P. Jaroniec, and Witold K. Surewicz. 2022. “Cryo-EM Structure of Disease-Related Prion Fibrils Provides Insights into Seeding Barriers.” Nature Structural & Molecular Biology 29 (10): 962–65. https://doi.org/10.1038/s41594-022-00833-4.
Mead, Simon, James Uphill, John Beck, Mark Poulter, Tracy Campbell, Jessica Lowe, Gary Adamson, et al. 2012. “Genome-Wide Association Study in Multiple Human Prion Diseases Suggests Genetic Risk Factors Additional to PRNP.” Human Molecular Genetics 21 (8): 1897–1906. https://doi.org/10.1093/hmg/ddr607.
Minikel, Eric Vallabh, Sonia M. Vallabh, Monkol Lek, Karol Estrada, Kaitlin E. Samocha, J. Fah Sathirapongsasuti, Cory Y. McLean, et al. 2016. “Quantifying Prion Disease Penetrance Using Large Population Control Cohorts.” Science Translational Medicine 8 (322): 322ra9. https://doi.org/10.1126/scitranslmed.aad5169.
Palmer, M. S., A. J. Dryden, J. T. Hughes, and J. Collinge. 1991. “Homozygous Prion Protein Genotype Predisposes to Sporadic Creutzfeldt-Jakob Disease.” Nature 352 (6333): 340–42. https://doi.org/10.1038/352340a0.
Pan, K. M., M. Baldwin, J. Nguyen, M. Gasset, A. Serban, D. Groth, I. Mehlhorn, Z. Huang, R. J. Fletterick, and F. E. Cohen. 1993. “Conversion of Alpha-Helices into Beta-Sheets Features in the Formation of the Scrapie Prion Proteins.” Proceedings of the National Academy of Sciences of the United States of America 90 (23): 10962–66. https://doi.org/10.1073/pnas.90.23.10962.
Prusiner, S. B. 1998. “Prions.” Proceedings of the National Academy of Sciences of the United States of America 95 (23): 13363–83. https://doi.org/10.1073/pnas.95.23.13363.
Sawaya, Michael R., Michael P. Hughes, Jose A. Rodriguez, Roland Riek, and David S. Eisenberg. 2021. “The Expanding Amyloid Family: Structure, Stability, Function, and Pathogenesis.” Cell 184 (19): 4857–73. https://doi.org/10.1016/j.cell.2021.08.013.
Tagliavini, F., F. Prelli, J. Ghiso, O. Bugiani, D. Serban, S. B. Prusiner, M. R. Farlow, B. Ghetti, and B. Frangione. 1991. “Amyloid Protein of Gerstmann-Sträussler-Scheinker Disease (Indiana Kindred) Is an 11 Kd Fragment of Prion Protein with an N-Terminal Glycine at Codon 58.” The EMBO Journal 10 (3): 513–19. https://doi.org/10.1002/j.1460-2075.1991.tb07977.x.
Wang, Li-Qiang, Kun Zhao, Han-Ye Yuan, Xiang-Ning Li, Hai-Bin Dang, Yeyang Ma, Qiang Wang, et al. 2021. “Genetic Prion Disease–Related Mutation E196K Displays a Novel Amyloid Fibril Structure Revealed by Cryo-EM.” Science Advances 7 (37): eabg9676. https://doi.org/10.1126/sciadv.abg9676.
Wang, Li-Qiang, Kun Zhao, Han-Ye Yuan, Qiang Wang, Zeyuan Guan, Jing Tao, Xiang-Ning Li, et al. 2020. “Cryo-EM Structure of an Amyloid Fibril Formed by Full-Length Human Prion Protein.” Nature Structural & Molecular Biology 27 (6): 598–602. https://doi.org/10.1038/s41594-020-0441-5.
Microtubule-associated protein tau ↓
Microtubule-associated protein tau, also simply called tau, is a neuronal protein whose main function is binding to and stabilizing microtubules(Goedert 2005). Tau is found in amyloid deposits in a plethora of neurodegenerative diseases, giving them the collective title of “tauopathies”(Goedert, Eisenberg, and Crowther 2017) and mutations in tau are connected to many of these diseases, which fall under the umbrella of diseases referred to as “frontotemporal dementia and parkinsonism linked to chromosome 17” (FTDP-17)(Goedert 2005; Wolfe 2009; Buée et al. 2000). Tau is an especially well-studied amyloid protein and many distinct fibril structures have been determined for tau. This has revealed the phenomenon of different diseases harboring distinct fibril polymorphs of tau(Shi et al. 2021), i.e. the molecular structure of tau fibrils are consistent between Alzheimer’s disease patients, but it is different from the molecular structure of tau fibrils from patients with chronic traumatic encephalopathy, while the structures of fibrils are consistent between people suffering from that disease, and so on for Pick’s disease, corticobasal degeneration (CBD), argyrophilic grain disease (AGD), etc.. The vast majority of these fibril structures come from protein without mutations, however, so the structural trends for mutant tau was not known. Also, structures have been determined from tau fibrils from individuals with disease-causing intronic mutations in the MAPT gene (unaltered protein sequence) which are identical to those from sporadic AGD(Shi et al. 2021). Recently, multiple mutant structures have been determined for tau fibrils: ΔK281 (pdb ID: 8p34)(Schweighauser, Garringer, et al. 2023), P301S in 0N4R tau transgenic mice (Tg2541) (pdb ID: 8q96)(Schweighauser, Murzin, et al. 2023), and P301S in 1N4R tau transgenic mice (PS19) (pdb ID: 8q92)(Schweighauser, Murzin, et al. 2023) (Supp. Fig. 3). These structures shed some light on how mutations affect the relationship between structure and disease.
The ΔK281 mutation causes exon 10 skipping(van Swieten et al. 2007) and causes frontotemporal dementia (FTD) with abundant Pick bodies (inclusions of 3R, but not 4R, tau). Consistent with this, the structure of tau fibrils extracted from individuals with this mutation are nearly identical to the structure of fibrils extracted from individuals with Pick’s disease (pdb ID: 6gx5)(Falcon et al. 2018) (Supp. Fig. 3a). The similarity of the two structures is so great that ΔK281 is considered to explicitly cause an inherited form of Pick’s disease(Schweighauser, Garringer, et al. 2023). The region of the mutation is not present in the fibril structure since K280 is within repeat 2 of tau which is not present in the 3R tau making up the fibrils. These findings evince the importance of 3R/4R tau ratios in disease, since recombinant 4R tau with a ΔK281 mutation does have increased aggregation propensity(Barghorn et al. 2000), but no 4R tau was found in the fibrils extracted from patient brain. Thus, this mutant fibril structure is the same as the structure of the protein in the sporadic form of the same disease.
The structures of P301S tau, however, are distinct from all other tau fibril structures thus determined as well as from each other (Supp. Fig. 3a). The P301S mutation causes FTDP-17 in humans, but is not a splicing mutation like ΔK281. It should be noted that the relevance of the differences between the two P301S structures to human tauopathies is difficult to gauge since the samples for structural studies were acquired from mouse models expressing human protein, not humans. Although the structures are quite different from each other, which could be due to the different forms of tau in each mouse or other differences in the characteristics of the mouse lines themselves, they both share a common substructure: a steric zipper formed by a dagger-shaped fold of residues D294-K311 (Supp. Fig. 3c). This substructure is also shared by wild-type tau structures determined from fibrils extracted from patients with limbic-predominant neuronal inclusion body 4R tauopathy (LNT) (pdb IDs: 7p6a, 7p6b, 7p6c)(Shi et al. 2021) (Supp. Fig. 3). This substructure also resembles the first intermediate amyloid (FIA) (pdb ID: 8ppo)(Lövestam et al. 2023), although the FIA is dimeric. The substructure present in the mutant fibrils may indicate a monomeric intermediate analogous to the FIA which is promoted by the P301S mutation, since the mutation creates an additional hydrogen bond which would stabilize the fold. Thus, the P301S mutation may stabilize a substructure which leads to an overall unique amyloid fold.
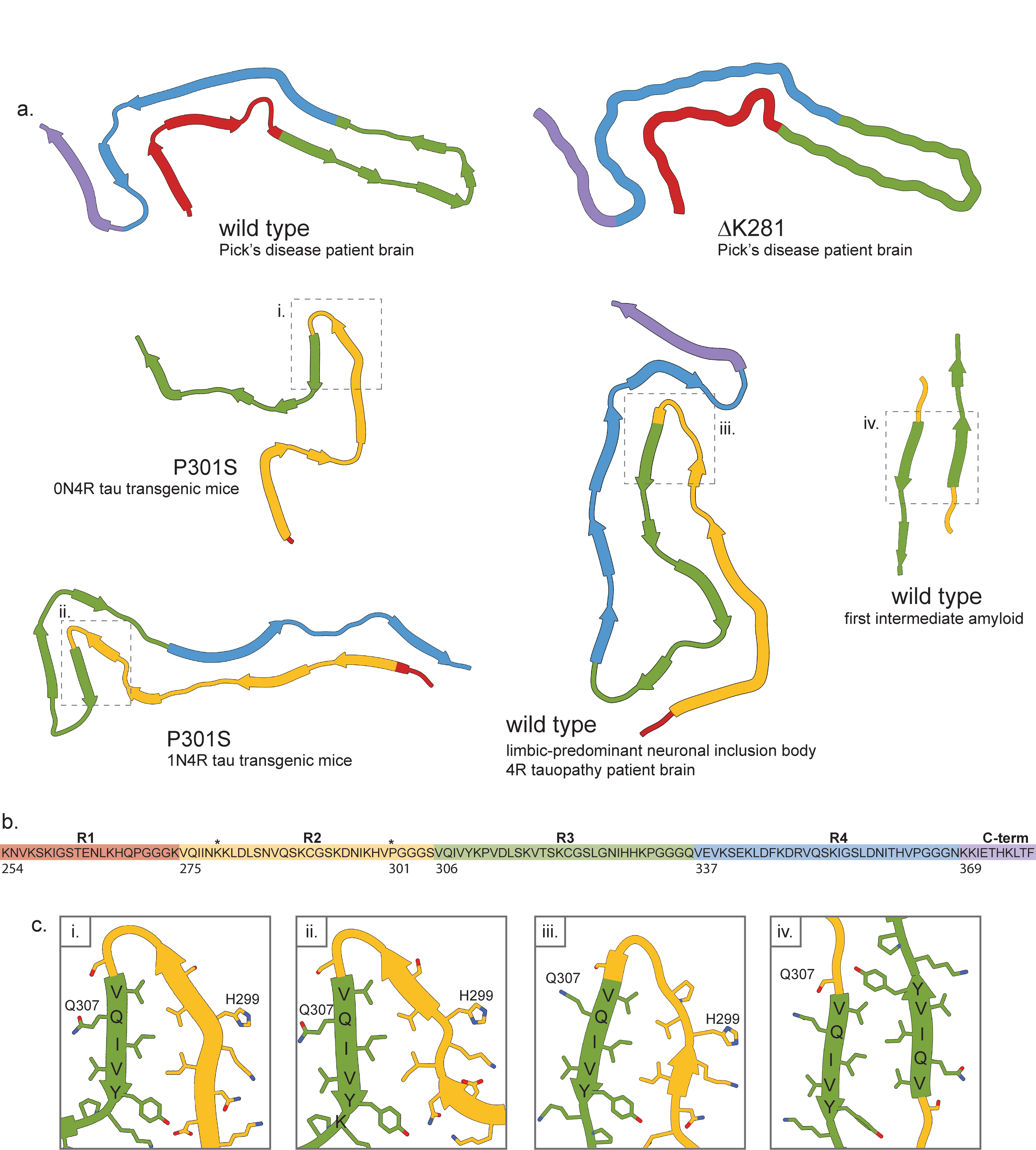
Supplemental Figure 3: Fibril structures of microtubule-associated protein tau. a) Structures of amyloid fibril cores of wild-type (Pick’s disease patient brain: pdb id 6gx5; limbic-predominant neuronal inclusion body 4R tauopathy patient brain: pdb id 7p6a; first intermediate filament: pdb id 8ppo), ΔK281 (pdb id 8p34), and P301S (top: pdb id 8q96; bottom: pdb id 8q92) microtubule-associated protein tau. b) Amino acid sequence of microtubule-associated protein tau represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). The fold common to both P301S structures and one of the wild-type structures is highlighted, as well as the aggregation-promoting amino acid sequence that is common to them and the first intermediate filament fibril core: VQIVYK.
Barghorn, S., Q. Zheng-Fischhöfer, M. Ackmann, J. Biernat, M. von Bergen, E. M. Mandelkow, and E. Mandelkow. 2000. “Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias.” Biochemistry 39 (38): 11714–21. https://doi.org/10.1021/bi000850r.
Buée, L., T. Bussière, V. Buée-Scherrer, A. Delacourte, and P. R. Hof. 2000. “Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders.” Brain Research. Brain Research Reviews 33 (1): 95–130. https://doi.org/10.1016/s0165-0173(00)00019-9.
Falcon, Benjamin, Wenjuan Zhang, Alexey G. Murzin, Garib Murshudov, Holly J. Garringer, Ruben Vidal, R. Anthony Crowther, Bernardino Ghetti, Sjors H. W. Scheres, and Michel Goedert. 2018. “Structures of Filaments from Pick’s Disease Reveal a Novel Tau Protein Fold.” Nature 561 (7721): 137–40. https://doi.org/10.1038/s41586-018-0454-y.
Goedert, Michel. 2005. “Tau Gene Mutations and Their Effects.” Movement Disorders: Official Journal of the Movement Disorder Society 20 Suppl 12 (August):S45-52. https://doi.org/10.1002/mds.20539.
Goedert, Michel, David S. Eisenberg, and R. Anthony Crowther. 2017. “Propagation of Tau Aggregates and Neurodegeneration.” Annual Review of Neuroscience 40 (July):189–210. https://doi.org/10.1146/annurev-neuro-072116-031153.
Lövestam, Sofia, David Li, Jane L. Wagstaff, Abhay Kotecha, Dari Kimanius, Stephen H. McLaughlin, Alexey G. Murzin, Stefan M. V. Freund, Michel Goedert, and Sjors H. W. Scheres. 2023. “Disease-Specific Tau Filaments Assemble via Polymorphic Intermediates.” bioRxiv. https://doi.org/10.1101/2023.07.24.550295.
Schweighauser, Manuel, Holly J. Garringer, Therése Klingstedt, K. Peter R. Nilsson, Masami Masuda-Suzukake, Jill R. Murrell, Shannon L. Risacher, et al. 2023. “Mutation ∆K281 in MAPT Causes Pick’s Disease.” Acta Neuropathologica 146 (2): 211–26. https://doi.org/10.1007/s00401-023-02598-6.
Schweighauser, Manuel, Alexey G. Murzin, Jennifer Macdonald, Isabelle Lavenir, R. Anthony Crowther, Sjors H. W. Scheres, and Michel Goedert. 2023. “Cryo-EM Structures of Tau Filaments from the Brains of Mice Transgenic for Human Mutant P301S Tau.” Acta Neuropathologica Communications 11 (1): 160. https://doi.org/10.1186/s40478-023-01658-y.
Shi, Yang, Wenjuan Zhang, Yang Yang, Alexey G. Murzin, Benjamin Falcon, Abhay Kotecha, Mike van Beers, et al. 2021. “Structure-Based Classification of Tauopathies.” Nature 598 (7880): 359–63. https://doi.org/10.1038/s41586-021-03911-7.
Swieten, John C. van, Iraad F. Bronner, Asma Azmani, Lies-Anne Severijnen, Wouter Kamphorst, Rivka Ravid, Patrizia Rizzu, Rob Willemsen, and Peter Heutink. 2007. “The DeltaK280 Mutation in MAP Tau Favors Exon 10 Skipping in Vivo.” Journal of Neuropathology and Experimental Neurology 66 (1): 17–25. https://doi.org/10.1097/nen.0b013e31802c39a4.
Wolfe, Michael S. 2009. “Tau Mutations in Neurodegenerative Diseases.” The Journal of Biological Chemistry 284 (10): 6021–25. https://doi.org/10.1074/jbc.R800013200.
TAR DNA-binding protein 43 ↓
TAR DNA-binding protein 43, like the heterogeneous ribonucleoproteins, has a functional aggregated state(Lagier-Tourenne, Polymenidou, and Cleveland 2010; Colombrita et al. 2009; Moisse et al. 2009; Nishimoto et al. 2010). Though not necessarily a functional amyloid, the protein is able to form reversible, potentially amyloid-like, aggregates(Cao et al. 2019). Disruption of its normal self-interaction within the nucleus seems to be a key mechanism of familial mutations(Conicella et al. 2016) and pathological inclusion of TAR DNA-binding protein 43 are found mislocalized in the cytoplasm(Lagier-Tourenne, Polymenidou, and Cleveland 2010; Johnson et al. 2009; Jiang et al. 2016; Neumann et al. 2006; Kwong et al. 2008; Jiang et al. 2013). Pathological inclusions of this protein are comprised of a C-terminal fragment consisting of residues spanning positions 252-414(Neumann et al. 2006). There are more than 60 known mutations in this protein associated with amyotrophic lateral sclerosis (ALS) or frontotemporal lobar dementia with TDP-43 inclusions (FTLD-TDP), but the only mutation for which we have the structure of an amyloid fibril is A315E (pdb ID: 6n3c)(Cao et al. 2019) (Supp. Fig. 4) and this structure was solved from fibrils generated from a short fragment of the protein spanning residues 286-331 and the structure itself resolves residues 288-319. This makes comparison to wild-type structures difficult because many of them are solved from fibrils made from a different segment of the protein (residues 311-360)(Cao et al. 2019) or much larger constructs such as the entire low-complexity domain (residues 267-414)(Li, Babinchak, and Surewicz 2021). Fortuitously, the wild-type structures with the most comparable sequence identity were solved from fibrils extracted from two individuals diagnosed with ALS with FTLD type B (both patients’ fibrils revealed the same structure) (pdb ID: 7py2)(Arseni et al. 2022) and from fibrils extracted from three individuals diagnosed with FTLD type A without ALS (all three patients’ fibrils revealed the same structure) (pdb ID: 8cg3)(Arseni et al. 2023) (Supp. Fig. 4). However, the comparison is still complicated by the fact that this patient-extracted structures still resolve significantly larger fragments of the protein than the mutant structure, residues 282-360 (type B) and residues 272-360 (type A) (Supp. Fig. 4a,b), and some of the differences in the structures may be accountable to the differing length of protein fragments forming the fibrils rather than the mutation itself. Also, the two patient-extracted wild-type structures are quite different from each other, consistent with the different disease origins.
With these considerations in mind, we can still identify structural changes that can likely be attributed to the mutation. The mutation itself, A315E, is unusual for amyloidogenic mutations because it is a transition from a nonpolar residue to a charged residue, while most amyloidogenic transitions go in the other direction(Rosenberg et al. 2022). This new charged residue is complementary to the nearby R293, which is solvent-facing in the type B wild-type structure and buried without an electrostatic interaction in the type A structure (Supp. Fig. 4c). In the mutant structure, E315 and R293 form an electrostatic interaction which requires a significant structural conversion relative to the wild-type structures and creates several new side chain interactions (Supp. Fig. 4c). In relation to the type B wild-type structure, one such interaction is L299 participating in hydrophobic interactions with M307, F313, and M311, whereas in the type B wild-type structure L299 is far away from those residues (Supp. Fig. 4c). In relation to the type A wild-type structure, the hydrophobic interaction of L299, M307, F313, and M311 is present in the wild-type structure, but in the mutant structure the interaction of E315 and R293 affects the positions of residues C-terminal to E315, such as exposing S317 to solvent, and residues N-terminal to R293, such as exposing N291 to solvent (Supp. Fig. 4c). Another, perhaps more obvious, difference is that the wild-type structure consists of a single protofilament while the mutant structure consists of four. It is unclear, however, if the high number of protofilaments in the mutant structure would form if a longer construct were used to form the fibrils, since interprotofilament interactions which are observed in the mutant structure are blocked by intrachain interactions in the type A wild-type fold. Regardless, the mutant structure with its multiple protofilaments creates a more stable fibril than the wild-type in regard to solvation energy.
It is worth noting that E315 may also interact with a different nearby arginine residue R361, in which case a completely different structure may form. However, this cannot be confirmed without structural characterization of either a longer construct of the protein or of fibrils extracted from a patient with the same mutation.
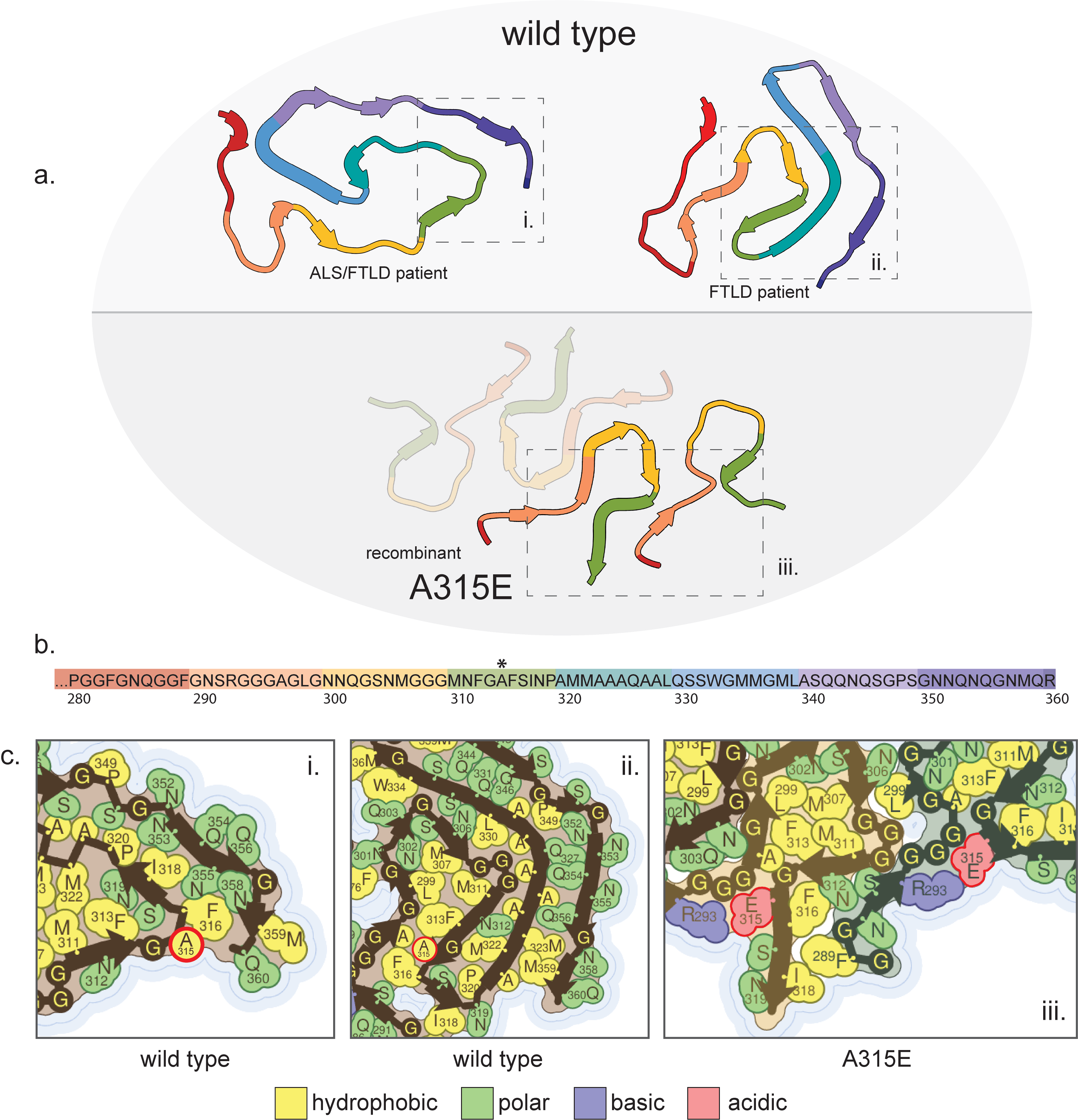
Supplemental Figure 4: Fibril structures of TAR DNA-binding protein 43. a) Structures of amyloid fibril cores of wild-type (left: pdb id 7py2; right: pdb id 8cg3) and A315E (pdb id 6n3c) TAR DNA-binding protein 43. b) Amino acid sequence of TAR DNA-binding protein 43 represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). Mutated residues are highlighted by a red outline.
Arseni, Diana, Renren Chen, Alexey G. Murzin, Sew Y. Peak-Chew, Holly J. Garringer, Kathy L. Newell, Fuyuki Kametani, et al. 2023. “TDP-43 Forms Amyloid Filaments with a Distinct Fold in Type A FTLD-TDP.” Nature 620 (7975): 898–903. https://doi.org/10.1038/s41586-023-06405-w.
Arseni, Diana, Masato Hasegawa, Alexey G. Murzin, Fuyuki Kametani, Makoto Arai, Mari Yoshida, and Benjamin Ryskeldi-Falcon. 2022. “Structure of Pathological TDP-43 Filaments from ALS with FTLD.” Nature 601 (7891): 139–43. https://doi.org/10.1038/s41586-021-04199-3.
Cao, Qin, David R. Boyer, Michael R. Sawaya, Peng Ge, and David S. Eisenberg. 2019. “Cryo-EM Structures of Four Polymorphic TDP-43 Amyloid Cores.” Nature Structural & Molecular Biology 26 (7): 619–27. https://doi.org/10.1038/s41594-019-0248-4.
Colombrita, Claudia, Eleonora Zennaro, Claudia Fallini, Markus Weber, Andreas Sommacal, Emanuele Buratti, Vincenzo Silani, and Antonia Ratti. 2009. “TDP-43 Is Recruited to Stress Granules in Conditions of Oxidative Insult.” Journal of Neurochemistry 111 (4): 1051–61. https://doi.org/10.1111/j.1471-4159.2009.06383.x.
Conicella, Alexander E., Gül H. Zerze, Jeetain Mittal, and Nicolas L. Fawzi. 2016. “ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain.” Structure (London, England: 1993) 24 (9): 1537–49. https://doi.org/10.1016/j.str.2016.07.007.
Jiang, Lei-Lei, Mei-Xia Che, Jian Zhao, Chen-Jie Zhou, Mu-Yun Xie, Hai-Yin Li, Jian-Hua He, and Hong-Yu Hu. 2013. “Structural Transformation of the Amyloidogenic Core Region of TDP-43 Protein Initiates Its Aggregation and Cytoplasmic Inclusion.” The Journal of Biological Chemistry 288 (27): 19614–24. https://doi.org/10.1074/jbc.M113.463828.
Jiang, Lei-Lei, Jian Zhao, Xiao-Fang Yin, Wen-Tian He, Hui Yang, Mei-Xia Che, and Hong-Yu Hu. 2016. “Two Mutations G335D and Q343R within the Amyloidogenic Core Region of TDP-43 Influence Its Aggregation and Inclusion Formation.” Scientific Reports 6 (March):23928. https://doi.org/10.1038/srep23928.
Johnson, Brian S., David Snead, Jonathan J. Lee, J. Michael McCaffery, James Shorter, and Aaron D. Gitler. 2009. “TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity.” The Journal of Biological Chemistry 284 (30): 20329–39. https://doi.org/10.1074/jbc.M109.010264.
Kwong, Linda K., Kunihiro Uryu, John Q. Trojanowski, and Virginia M.-Y. Lee. 2008. “TDP-43 Proteinopathies: Neurodegenerative Protein Misfolding Diseases without Amyloidosis.” Neuro-Signals 16 (1): 41–51. https://doi.org/10.1159/000109758.
Lagier-Tourenne, Clotilde, Magdalini Polymenidou, and Don W. Cleveland. 2010. “TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration.” Human Molecular Genetics 19 (R1): R46-64. https://doi.org/10.1093/hmg/ddq137.
Li, Qiuye, W. Michael Babinchak, and Witold K. Surewicz. 2021. “Cryo-EM Structure of Amyloid Fibrils Formed by the Entire Low Complexity Domain of TDP-43.” Nature Communications 12 (1): 1620. https://doi.org/10.1038/s41467-021-21912-y.
Moisse, Katie, Kathryn Volkening, Cheryl Leystra-Lantz, Ian Welch, Tracy Hill, and Michael J. Strong. 2009. “Divergent Patterns of Cytosolic TDP-43 and Neuronal Progranulin Expression Following Axotomy: Implications for TDP-43 in the Physiological Response to Neuronal Injury.” Brain Research 1249 (January):202–11. https://doi.org/10.1016/j.brainres.2008.10.021.
Neumann, Manuela, Deepak M. Sampathu, Linda K. Kwong, Adam C. Truax, Matthew C. Micsenyi, Thomas T. Chou, Jennifer Bruce, et al. 2006. “Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis.” Science (New York, N.Y.) 314 (5796): 130–33. https://doi.org/10.1126/science.1134108.
Nishimoto, Yoshinori, Daisuke Ito, Takuya Yagi, Yoshihiro Nihei, Yoshiko Tsunoda, and Norihiro Suzuki. 2010. “Characterization of Alternative Isoforms and Inclusion Body of the TAR DNA-Binding Protein-43.” The Journal of Biological Chemistry 285 (1): 608–19. https://doi.org/10.1074/jbc.M109.022012.
Rosenberg, Gregory M., Kevin A. Murray, Lukasz Salwinski, Michael P. Hughes, Romany Abskharon, and David S. Eisenberg. 2022. “Bioinformatic Identification of Previously Unrecognized Amyloidogenic Proteins.” The Journal of Biological Chemistry 298 (5): 101920. https://doi.org/10.1016/j.jbc.2022.101920.
α-synuclein ↓
α-synuclein is a protein whose function is not entirely clear, but localizes to presynaptic terminals and interacts with lipid membranes, i.e. vesicles, and is able to adopt an α-helical secondary structure when associated with membranes despite being disordered in solution(Burré 2015). This protein is the main component of Lewy bodies and Lewy neurites which are the hallmarks of Parkinson’s disease (PD) and dementia with Lewy bodies (DLB)(Spillantini et al. 1997) and also aggregates in multiple system atrophy (MSA) as well as other “synucleinopathies”. Mutations in this protein cause early onset of these diseases and we have amyloid structural data for five mutations (only one mutant structure is from patient-extracted fibrils: T22TMAAAEKT) as well as many wild-type structures including some extracted from patients with MSA or PD and DLB. The mutations for which we have structures are H50Q (pdb IDs: 6peo,6pes)(Boyer et al. 2019), A53T (pdb ID: 6lrq)(Y. Sun et al. 2020), A53E (pdb ID: 7uak)(C. Sun et al. 2023), E46K (pdb IDs: 6l4s,6ufr)(Zhao et al. 2020; Boyer et al. 2020), G51D(pdb ID: 7e0f)(Y. Sun et al. 2021), and T22TMAAAEKT (pdb IDs: 8bqv,8bqw)(Yang et al. 2023) (Supp. Fig. 5). When comparing structures, we must keep in mind that different diseases caused by α-synuclein harbor distinct amyloid folds, so differences in mutant structures should be viewed in light of this fact. This is complicated by the fact that all but one of the mutations listed above (T22TMAAAEKT being the exception) are associated with early-onset PD or DLB, but the protofilament folds of the H50Q and A53T mutant fibrils seem to be more similar to the MSA brain fold (pdb IDs: 6xyq, 6xyp,6xyo)(Schweighauser et al. 2020) than the PD and DLB brain fold (pdb ID: 8a9l)(Yang et al. 2022) (Supp. Fig. 5), these being the patient-extracted wild-type structures mentioned above. Also, the patient-extracted fibril structures for both MSA and PD/DLB, despite distinct folds, show extra non-peptide density coordinated mainly by lysine residues (K43, K45, and H50 of both protofilaments in MSA; K32, K34, K43, K45, and Y39 of the single protofilament in PD) (Supp. Fig. 5c), but it is not known what cofactor creates this density. The presence of this cofactor may be important for the formation of these specific patient structures, and all recombinant structures must be presumably qualified by the exclusion of this unknown cofactor.
The H50Q mutant α-synuclein forms a fibril with a very similar fold to recombinant wild-type structures (pdb IDs: 6cu7,6cu8)(Li et al. 2018) and the patient-extracted MSA fold (also wild-type), but has a distinct protofilament interface or, in one isoform, only a single protofilament (Supp. Fig. 5). The mutation may contribute to this altered interface by abolishing a potential electrostatic interaction between H50 and E57, as seen in the 6cu7 structure, and instead Q50 forms an intramolecular hydrogen bond with K45(Boyer et al. 2019) (Supp. Fig. 5c). Q50 may also induce a steric clash with E57. However, based on this structure, the stabilities of the mutant and wild-type structures are comparable to each other. Although, denaturation resistance assays establish a higher stability of H50Q fibrils than wild-type(Porcari et al. 2015), so the structure of the fibril core may not be the only contributor to overall stability. It is unclear why this mutation favors a fold more similar to the MSA patient fold than the PD patient fold.
The A53T mutant forms a fibril which is extremely similar to those formed by the H50Q mutant, although superstructural features, like overall fibril twist, differ(Y. Sun et al. 2020) (Supp. Fig. 5a). In some recombinant wild-type structures, e.g. 6cu7, A53 is at the center of the protofilament interface (Supp. Fig. 5c) and the A53T mutation directly disrupts this interface through a steric clash, favoring the interface also seen formed by the H50Q mutant protein. Again, this interface appears less stable than the wild-type interface, making the increased aggregation propensity of the mutant protein(Y. Sun et al. 2020; Narhi et al. 1999; Conway, Harper, and Lansbury 1998) difficult to explain by the structure of the resulting fibril alone. And, again, it is unclear why this mutation favors a fold more similar to the MSA patient fold than the PD patient fold.
The A53E mutant forms a fibril which is extremely similar to the A53T fibril, with only some slight differences in the packing of some side chains (Supp. Fig. 5a). Like the A53T mutant, the A53E mutant introduces a steric clash into the wild-type protofilament interface. However, unlike A53T, the A53E mutant slows fibril formation relative to the wild-type protein(C. Sun et al. 2023). This may be due to slight structural variations that seems to make the already small protofilament interface which is common between both the A53T and A53E mutant fibrils even less stable in the A53E fibril(C. Sun et al. 2023), although it is unclear how the mutation contributes to this difference. It is also unclear how this mutation contributes to disease, but it should be noted that a difference from the wild-type fibril which is shared between the H50Q mutant, A53T mutant, and A53E mutant is the inversion of residue K58 from outward-facing (wild-type) to inward-facing (mutants). This change compacts the interior of the protofilament core, which excludes potential cofactors and may make the protofilament fold more favorable in relation to the wild-type.
Two distinct structures have been solved for fibrils with the E46K mutation. These two structures even have apparently different biochemical properties, with the 6l4s fibrils being apparently less stable than wild-type fibrils(Zhao et al. 2020) while the 6ufr fibrils are apparently more stable than wild-type fibrils(Boyer et al. 2020). However, both structures represent a departure from the fold observed in the H50Q mutant structure, the A53T mutant structure, the A53E mutant structure, some recombinant wild-type structures, and the patient-extracted MSA structure (Supp. Fig. 5a). The E46K mutation disrupts a salt-bridge formed by E46 and K80 that is present in the aforementioned structures. In the E46K structures, the removal of this electrostatic interaction results in a complete rearrangement of the core. However, the PD and DLB patient-extracted structure does not contain the E46-K80 interaction, despite not having this mutation, and has a distinct fold to both E46K structures. So, the particular arrangement of the fibril core seen in these recombinant mutant structures cannot be entirely attributable to the mutation, especially since the mutated residue does not seem to participate in any structure-specific interactions. The differences between the two mutant structures may be attributable to differences in the preparation of fibrils, i.e. different buffers, different protein concentrations, and the 6l4s fibrils were grown in the presence of preformed fibril seeds. And, again, neither structure was obtained from fibrils grown in the presence of whatever cofactor creates the density in the 8a9l and 6xyq/6xyp/6xyo structures. The coordination of lysines around this cofactor may preclude the K45-E57 interactions seen in both E46K structures. In all, the mutation explains certain structural differences from the human-extracted MSA structure as well as recombinant wild-type structures bearing an E46-K80 interaction, but cannot explain the differences from the wild-type PD and DLB structure.
The G51D mutant structure is extremely similar to the 6l4s E46K structure, both fibrils even bearing a unique right-handed twist(Y. Sun et al. 2021), although D51 abolishes a turn present in the E46K structure which allows the interaction between K45 and E57 (Supp. Fig. 5). Also, like the 6l4s fibrils, the G51D fibrils were biochemically shown to be less stable than wild-type fibrils, but also more cytotoxic and more seeding-competent(Y. Sun et al. 2021). A decrease in stability has been hypothesized to increase propagation of seeds for other mutations as well(Y. Sun et al. 2020; Zhao et al. 2020). Like the A53T mutation, the G51D mutation creates a steric clash that would directly disrupt the protofilament interface of some recombinant wild-type fibrils, but it is not entirely clear why these two mutations induce such distinct structures from each other. The G51D mutation may also be somewhat incompatible with the patient-extracted PD structure, since it could form a steric clash with the undefined peptide density interacting with the H50-V55 region, but it is unclear how important this interface is to the overall structure of that fibril core. Also, none of the lysines coordinating the mystery density in the patient-derived structure are resolved in the G51D structure, so it is unclear if they could be part of the ordered core if the cofactor were present or if D51 somehow precludes this interaction.
The T22TMAAAEKT mutation is a 7 amino acid insertion after T22 which was found in a patient with extremely early onset of disease (13 years old) and rapid progression (2 years from manifestation of symptoms to death)(Yang et al. 2023; Takao et al. 2004). This was the only case of a disease now known as juvenile-onset synucleinopathy, so named distinctly from DLB due to the discovery of the unique fold of α-synuclein fibrils extracted from the patient’s brain. The fibril consists of a single protofilament with a fold very similar to the patient-extracted MSA fibril, containing the salt bridge formed from E46 and K80 (Supp. Fig. 5). The difference is that the protofilament interface is supplanted with an interaction with the N-terminal region of the protein harboring the insertion mutation. Like the other patient-extracted structures, this structure also contains a network of lysines coordinating an undefined density. The peptide density which forms the interface with H50-E57 is actually too ambiguous to definitively model in the mutant sequence, but the extra length of the mutant sequence is better able to fill in the other island of ambiguous peptide density forming an interface with K60-T64. It is also possible that the fibril contains both mutant and wild-type protein, leading to the ambiguity in the cryo-em data. The mutation probably contributes to this unique fold by making the intra-protofilament interfaces revealed in this structure easier to form by lengthening the protein with a repeated sequence. This mutation clearly induces a particularly aggressive synucleinopathy, but the structure does not immediately allow us to intuit why.
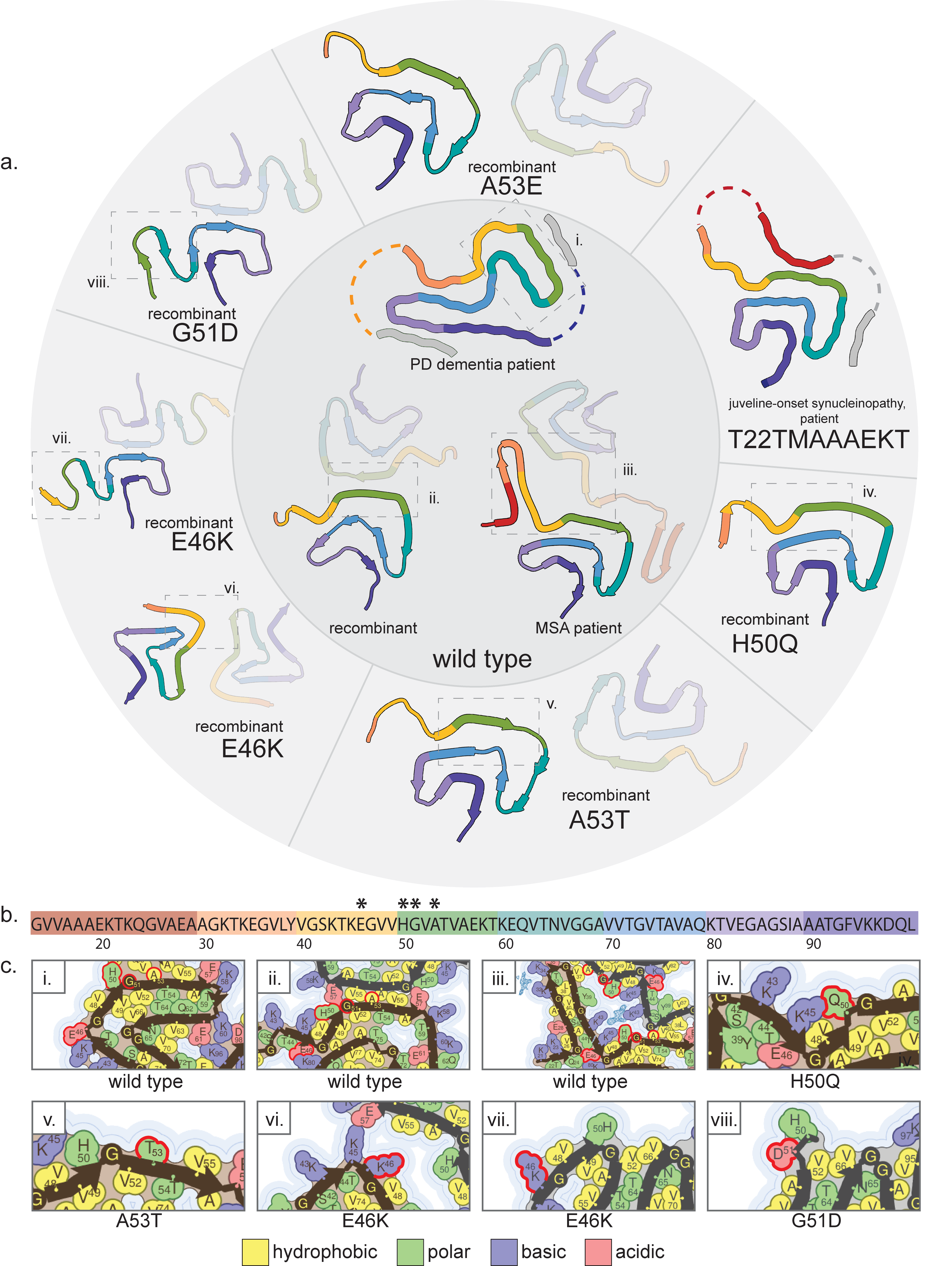
Supplemental Figure 5: Fibril structures of α-synuclein. a) Structures of amyloid fibril cores of wild-type (top: pdb id 8a9l; bottom left: pdb id 6cu7; bottom right: pdb id 6xyo), A53E (pdb id 7uak), T22TMAAAEKT (pdb id 8bqv), H50Q (pdb id 6peo), A53T (pdb id 6lrq), E46K (top: pdb id 6l4s; bottom: pdb id 6ufr), and G51D (pdb id 7e0f) α-synuclein. b) Amino acid sequence of α-synuclein represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). Mutated residues are highlighted by a red outline.
Boyer, David R., Binsen Li, Chuanqi Sun, Weijia Fan, Michael R. Sawaya, Lin Jiang, and David S. Eisenberg. 2019. “Structures of Fibrils Formed by α-Synuclein Hereditary Disease Mutant H50Q Reveal New Polymorphs.” Nature Structural & Molecular Biology 26 (11): 1044–52. https://doi.org/10.1038/s41594-019-0322-y.
Boyer, David R., Binsen Li, Chuanqi Sun, Weijia Fan, Kang Zhou, Michael P. Hughes, Michael R. Sawaya, Lin Jiang, and David S. Eisenberg. 2020. “The α-Synuclein Hereditary Mutation E46K Unlocks a More Stable, Pathogenic Fibril Structure.” Proceedings of the National Academy of Sciences of the United States of America 117 (7): 3592–3602. https://doi.org/10.1073/pnas.1917914117.
Burré, Jacqueline. 2015. “The Synaptic Function of α-Synuclein.” Journal of Parkinson’s Disease 5 (4): 699–713. https://doi.org/10.3233/JPD-150642.
Conway, K. A., J. D. Harper, and P. T. Lansbury. 1998. “Accelerated in Vitro Fibril Formation by a Mutant Alpha-Synuclein Linked to Early-Onset Parkinson Disease.” Nature Medicine 4 (11): 1318–20. https://doi.org/10.1038/3311.
Li, Binsen, Peng Ge, Kevin A. Murray, Phorum Sheth, Meng Zhang, Gayatri Nair, Michael R. Sawaya, et al. 2018. “Cryo-EM of Full-Length α-Synuclein Reveals Fibril Polymorphs with a Common Structural Kernel.” Nature Communications 9 (1): 3609. https://doi.org/10.1038/s41467-018-05971-2.
Narhi, L., S. J. Wood, S. Steavenson, Y. Jiang, G. M. Wu, D. Anafi, S. A. Kaufman, et al. 1999. “Both Familial Parkinson’s Disease Mutations Accelerate Alpha-Synuclein Aggregation.” The Journal of Biological Chemistry 274 (14): 9843–46. https://doi.org/10.1074/jbc.274.14.9843.
Porcari, Riccardo, Christos Proukakis, Christopher A. Waudby, Benedetta Bolognesi, P. Patrizia Mangione, Jack F. S. Paton, Stephen Mullin, et al. 2015. “The H50Q Mutation Induces a 10-Fold Decrease in the Solubility of α-Synuclein.” The Journal of Biological Chemistry 290 (4): 2395–2404. https://doi.org/10.1074/jbc.M114.610527.
Schweighauser, Manuel, Yang Shi, Airi Tarutani, Fuyuki Kametani, Alexey G. Murzin, Bernardino Ghetti, Tomoyasu Matsubara, et al. 2020. “Structures of α-Synuclein Filaments from Multiple System Atrophy.” Nature 585 (7825): 464–69. https://doi.org/10.1038/s41586-020-2317-6.
Spillantini, M. G., M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes, and M. Goedert. 1997. “Alpha-Synuclein in Lewy Bodies.” Nature 388 (6645): 839–40. https://doi.org/10.1038/42166.
Sun, Chuanqi, Kang Zhou, Peter DePaola, Woo Shik Shin, Trae Hillyer, Michael R. Sawaya, Ruowei Zhu, Chao Peng, Z. Hong Zhou, and Lin Jiang. 2023. “Cryo-EM Structure of Amyloid Fibril Formed by α-Synuclein Hereditary A53E Mutation Reveals a Distinct Protofilament Interface.” The Journal of Biological Chemistry 299 (4): 104566. https://doi.org/10.1016/j.jbc.2023.104566.
Sun, Yunpeng, Shouqiao Hou, Kun Zhao, Houfang Long, Zhenying Liu, Jing Gao, Yaoyang Zhang, Xiao-Dong Su, Dan Li, and Cong Liu. 2020. “Cryo-EM Structure of Full-Length α-Synuclein Amyloid Fibril with Parkinson’s Disease Familial A53T Mutation.” Cell Research 30 (4): 360–62. https://doi.org/10.1038/s41422-020-0299-4.
Sun, Yunpeng, Houfang Long, Wencheng Xia, Kun Wang, Xia Zhang, Bo Sun, Qin Cao, et al. 2021. “The Hereditary Mutation G51D Unlocks a Distinct Fibril Strain Transmissible to Wild-Type α-Synuclein.” Nature Communications 12 (1): 6252. https://doi.org/10.1038/s41467-021-26433-2.
Takao, Masaki, Bernardino Ghetti, Hirotaka Yoshida, Pedro Piccardo, Yolanda Narain, Jill R. Murrell, Ruben Vidal, et al. 2004. “Early-Onset Dementia with Lewy Bodies.” Brain Pathology (Zurich, Switzerland) 14 (2): 137–47. https://doi.org/10.1111/j.1750-3639.2004.tb00046.x.
Yang, Yang, Holly J. Garringer, Yang Shi, Sofia Lövestam, Sew Peak-Chew, Xianjun Zhang, Abhay Kotecha, et al. 2023. “New SNCA Mutation and Structures of α-Synuclein Filaments from Juvenile-Onset Synucleinopathy.” Acta Neuropathologica 145 (5): 561–72. https://doi.org/10.1007/s00401-023-02550-8.
Yang, Yang, Yang Shi, Manuel Schweighauser, Xianjun Zhang, Abhay Kotecha, Alexey G. Murzin, Holly J. Garringer, et al. 2022. “Structures of α-Synuclein Filaments from Human Brains with Lewy Pathology.” Nature 610 (7933): 791–95. https://doi.org/10.1038/s41586-022-05319-3.
Zhao, Kun, Yaowang Li, Zhenying Liu, Houfang Long, Chunyu Zhao, Feng Luo, Yunpeng Sun, et al. 2020. “Parkinson’s Disease Associated Mutation E46K of α-Synuclein Triggers the Formation of a Distinct Fibril Structure.” Nature Communications 11 (1): 2643. https://doi.org/10.1038/s41467-020-16386-3.
β2-microglobulin ↓
β2-microglobulin is a component of the class 1 major histocompatibility complex. It is shed by lymphocytes and circulating protein is usually filtered by the kidneys(Bernier 1980). It aggregates in patients receiving haemodialysis treatment due to increased plasma concentration of the protein, leading to a condition known as dialysis-related amyloidosis (DRA)(Gejyo et al. 1986; Dember and Jaber 2006). In addition, mutations can promote DRA and affect its clinical presentation (V47M)(Mizuno et al. 2021) as well as cause amyloidosis in absence of haemodialysis treatment (D96N, P52L)(Valleix et al. 2012; Prokaeva et al. 2022). We have structural data for fibrils formed by both V47M (pdb ID: 8a7q)(Wilkinson et al. 2023) and D96N (pdb ID: 8a7t)(Wilkinson et al. 2023) β2-microglobulin, as well as a truncated version of the protein commonly found in ex vivo fibrils, ΔN6 (pdb IDs: 8a7o, 8a7p)(Wilkinson et al. 2023) (Supp. Fig. 6). Wild-type β2-microglobulin (pdb ID: 6gk3)(Iadanza et al. 2018) (Supp. Fig. 6) does not fibrillize in vitro while close to neutral pH without additives which promote unfolding; the fibrils of the wild-type protein for which a structure was determined were formed at pH 2.5. Mutant β2-microglobulin, however, can form fibrils at only mildly acidic pH, 6.2, which mimics the environment of the arthritic joint(Wilkinson et al. 2023). Thus, many of the main differences in fibril structure between wild-type and mutant protein are reflective of stability at near-neutral pH.
Despite differences in protofilament interfaces and extensions of C- and/or N-termini, all of the mutant protofilaments (including ΔN6) have a common “hammer-shaped fold” which distinguishes them from the wild-type fibril protofilaments, while retaining the crucial disulfide bond between C80 and C25 (Supp. Fig. 6a,c). Both mutations (V47M and D96N) are located within the “head” of the hammer and thus likely contribute to the common shape of the protofilament fold, although it is unclear how the ΔN6 truncation promotes this shape as well. The distinct hammer head shape opens a solvent cavity within the protofilament core allowing for the solvation of several polar or charged residues in that region of the protein including D/N96. This may be promoted by the V47M mutation due to a steric clash of the methionine causing it to flip out toward solvent, thus enriching the region for polar side chains (Supp. Fig. 6c). The D96N mutation may promote this shape due to alleviation of the like charges of E94 and D96, allowing them both to point inward toward a solvent channel (Supp. Fig. 6c). The wild-type sequence, meanwhile, can likely only form at low pH since, in the wild-type fold, acidic side chains (E and D) have a highly disfavored orientation (pointing inward, away from solvent) if they are charged, which is the case at near-neutral pH (Supp. Fig. 6c). Thus, the common hammer-shaped fold of the mutant protein fibrils allows stability of the fibrils at more physiological pH compared to the wild-type sequence, which promotes fibril formation.
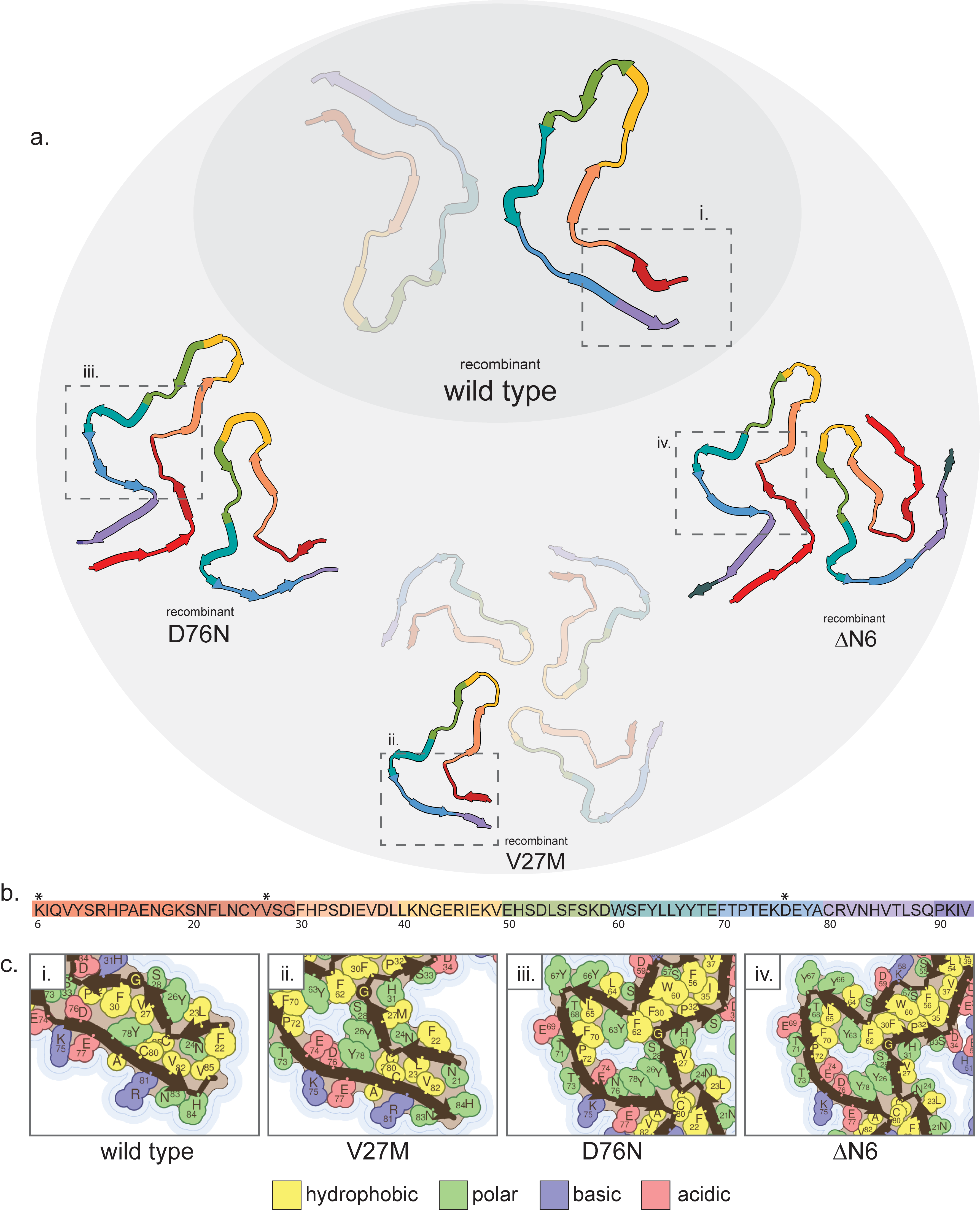
Supplemental Figure 6: Fibril structures of β2-microglobulin. a) Structures of amyloid fibril cores of wild-type (pdb id 6gk3), D76N (pdb id 8a7t), V27M (pdb id 8a7q), and ΔN6 (pdb id 8a7o) β2-microglobulin. ΔN6 is a fragment formed by proteolytic cleavage rather than a mutation, but it is included for its similarity to mutant structures. b) Amino acid sequence of β2-microglobulin represented in the fibril cores. Asterisks indicate locations of amyloidogenic mutations which are present in the structures reviewed here. c) Side chain details from the numbered boxes in a). Mutated residues are highlighted by a red outline.
Bernier, G. M. 1980. “Beta 2-Microglobulin: Structure, Function and Significance.” Vox Sanguinis 38 (6): 323–27. https://doi.org/10.1111/j.1423-0410.1980.tb04500.x.
Dember, Laura M., and Bertrand L. Jaber. 2006. “Dialysis-Related Amyloidosis: Late Finding or Hidden Epidemic?” Seminars in Dialysis 19 (2): 105–9. https://doi.org/10.1111/j.1525-139X.2006.00134.x.
Gejyo, F., S. Odani, T. Yamada, N. Honma, H. Saito, Y. Suzuki, Y. Nakagawa, H. Kobayashi, Y. Maruyama, and Y. Hirasawa. 1986. “Beta 2-Microglobulin: A New Form of Amyloid Protein Associated with Chronic Hemodialysis.” Kidney International 30 (3): 385–90. https://doi.org/10.1038/ki.1986.196.
Iadanza, Matthew G., Robert Silvers, Joshua Boardman, Hugh I. Smith, Theodoros K. Karamanos, Galia T. Debelouchina, Yongchao Su, Robert G. Griffin, Neil A. Ranson, and Sheena E. Radford. 2018. “The Structure of a Β2-Microglobulin Fibril Suggests a Molecular Basis for Its Amyloid Polymorphism.” Nature Communications 9 (1): 4517. https://doi.org/10.1038/s41467-018-06761-6.
Mizuno, Hiroki, Junichi Hoshino, Masatomo So, Yuta Kogure, Takeshi Fujii, Yoshifumi Ubara, Kenmei Takaichi, et al. 2021. “Dialysis-Related Amyloidosis Associated with a Novel Β2-Microglobulin Variant.” Amyloid: The International Journal of Experimental and Clinical Investigation: The Official Journal of the International Society of Amyloidosis 28 (1): 42–49. https://doi.org/10.1080/13506129.2020.1813097.
Prokaeva, Tatiana, Tracy Joshi, Elena S. Klimtchuk, Victoria M. Gibson, Brian Spencer, Omar Siddiqi, Dobrin Nedelkov, et al. 2022. “A Novel Substitution of Proline (P32L) Destabilises Β2-Microglobulin Inducing Hereditary Systemic Amyloidosis.” Amyloid: The International Journal of Experimental and Clinical Investigation: The Official Journal of the International Society of Amyloidosis 29 (4): 255–62. https://doi.org/10.1080/13506129.2022.2072199.
Valleix, Sophie, Julian D. Gillmore, Frank Bridoux, Palma P. Mangione, Ahmet Dogan, Brigitte Nedelec, Mathieu Boimard, et al. 2012. “Hereditary Systemic Amyloidosis Due to Asp76Asn Variant Β2-Microglobulin.” The New England Journal of Medicine 366 (24): 2276–83. https://doi.org/10.1056/NEJMoa1201356.
Wilkinson, Martin, Rodrigo U. Gallardo, Roberto Maya Martinez, Nicolas Guthertz, Masatomo So, Liam D. Aubrey, Sheena E. Radford, and Neil A. Ranson. 2023. “Disease-Relevant Β2-Microglobulin Variants Share a Common Amyloid Fold.” Nature Communications 14 (1): 1190. https://doi.org/10.1038/s41467-023-36791-8.