
David Eisenberg and his research group focus on protein interactions. In their experiments they study the structural basis for conversion of normal proteins to the amyloid state and conversion of prions to the infectious state. In computational work, they derive information on protein interactions from genomic and proteomic data, and design inhibitors of amyloid toxicity.
Amyloid and prion diseases are diseases of protein aggregation in which a normal, functional protein converts to an abnormal, aggregated protein. The systemic amyloid diseases, such as dialysis-related amyloidosis, are apparently caused by the accumulation of fibers until organs fail. The neurodegenerative amyloid diseases, such as Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis (ALS), and the prion conditions, seem to be caused by smaller oligomers, intermediate in size between monomers and fibers. Our goals are to understand the general features of the conversion to the amyloid state, why some of the diseases are transmissible between organisms and others not, what the structures of the toxic units are, and how they exert their toxic actions.
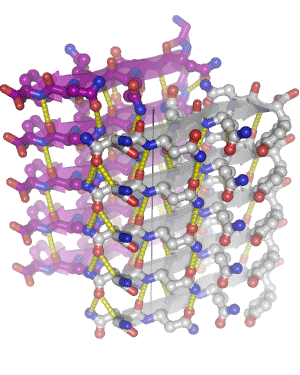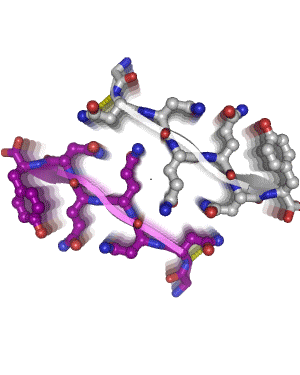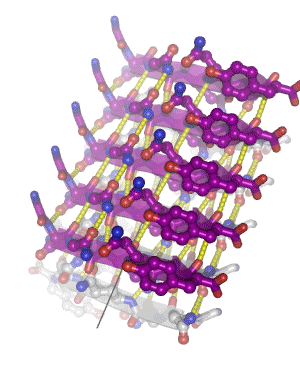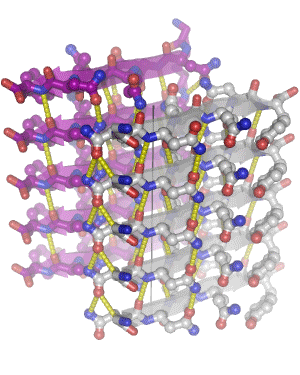
In 2005, we determined the atomic-level structure for the spine of an amyloid fiber. This structure shows that the spine consists of two parallel beta sheets, packed across a tight, dry interface that we call a steric zipper. The structure of the spine explains the stability of amyloid, gives hints about the conversion process, and suggests why some proteins form amyloid while others do not. Since 2005, we have determined some 160 amyloid spines from 15 disease-related proteins, using a combination of bioinformatics and structural tools. In 2010, we determined the structure of a small toxic amyloid-related oligomer, consisting of six anti-parallel beta strands forming a cylindrical barrel. This structure may suggest models for the toxic oligomers associated with amyloid diseases.

2015 Lab Picture
